1.本发明属于有机合成药物技术领域,具体涉及一种吲哚美辛的制备方法。
背景技术:
2.吲哚美辛是一种作用较强的解热镇痛剂,通过抑制环氧酶减少前列腺素(pg)合成而产生解热、镇痛及消炎作用,临床可用于急、慢性风湿性关节炎、痛风性关节炎及癌性疼痛;也用于滑囊炎、腱鞘炎及关节囊炎等;还可用于癌症引起的发热或其他难以控制的发热;也可用于抗血小板聚。
3.吲哚美辛的合成方法,文献报告的主要有三种。tsung-ying,shen等在专利us3285908a报道的合成路线如下:
[0004][0005]
kevin r.campos等人在organic letters 2004,vol6,no.1 79~82报道的合成路线如下:
[0006][0007]
i.v.magedov等人在chemistry of heterocyclic compounds(new york,ny,united states),41(4),449-451;2005报道的合成路线如下:
[0008][0009]
目前吲哚美辛的制备方法主要还存在以下问题:(1)以对甲氧基苯肼为原料,经乙醛保护,酰化,然后和乙酰丙酸在酸作用下发生关环反应得到吲哚美辛。中间体不稳定,不符合gmp质量控制要求;(2)以4-chloro-n-(p-tolyl)benzohydrazide为原料和当归内酯在酸作用下发生关环反应得到吲哚美辛,原料当归内酯没有商业化来源,而且价格是乙酰丙酸的30-40倍;(3)操作工艺复杂,不适于大规模工业化生产;(4)以对甲氧基苯胺为原料和亚硝酸钠在酸作用下生成重氮盐,然后经亚硫酸钠还原得到对甲氧基苯肼磺酸钠;对甲氧基苯肼磺酸钠酰化反应后和乙酰丙酸在酸作用下发生关环反应得到吲哚美辛。中间体不容易纯化,不符合gmp质量控制要求。
技术实现要素:
[0010]
本发明针对现有技术存在的问题,提供了一种吲哚美辛的制备方法。该方法操作简单,原料廉价易得,中间体稳定质量可控,符合gmp生产要求。
[0011]
为实现上述发明目的,本发明技术方案如下:
[0012]
一种吲哚美辛的制备方法,反应路线如下:
[0013][0014]
其中,r选自h、取代或未取代的烷基、取代或未取代的烯基、取代或未取代的炔基、取代或未取代的羰基、取代或未取代的环烷基、取代或未取代的环烯基、取代或未取代的芳基、取代或未取代的杂环基、取代或未取代的杂芳基或卤素。
[0015]
优选地,r选自h、取代或未取代的c1-c10烷基、取代或未取代的c2-c10烯基、取代或未取代的c2-c10炔基、取代或未取代的c1-c10羰基、取代或未取代的c3-c10环烷基、取代或未取代的c3-c10环烯基、取代或未取代的c6-c12芳基、取代或未取代的c5-c12杂环基、取
代或未取代的c5-c12杂芳基或卤素;
[0016]
所述取代的取代基选自卤素、硝基、氰基、氨基、羟基、羟甲基、羟乙基、巯基、羧基、酯基、芳基、杂环基、c1-c10烷基单取代胺基、c1-c10烷基双取代胺基、c1-c10烷氧基、c1-c10烷基羰氧基、c1-c10环烷基羰氧基、杂环基羰氧基、c1-c10烷氧基羰基、c1-c10环烷氧基羰基、杂环氧基羰基、c1-c10烷基羰胺基、c1-c10环烷基羰胺基、杂环基羰胺基、胺基羰基、c1-c10烷氧甲酰胺基、c1-c10烷巯基、羟基烷氧基、糖残基、磺酸基、磷酸基、多羟基c1-c10烷氧基羰基、羧基c1-c10烷氧基、羧基c1-c10烷基甲酰氧基中的一个或多个中的一个或多个。
[0017]
进一步优选地,r选自h、取代或未取代的c1-c8烷基、取代或未取代的c2-c8烯基、取代或未取代的c2-c8炔基、取代或未取代的c1-c8羰基、取代或未取代的c3-c8环烷基、取代或未取代的c3-c8环烯基、取代或未取代的c6-c10芳基、取代或未取代的c5-c10杂环基、取代或未取代的c5-c10杂芳基或卤素;
[0018]
所述取代的取代基选自卤素、硝基、氰基、氨基、羟基、羟甲基、羟乙基、巯基、羧基、酯基、苯基、杂环基、c1-c6烷基单取代胺基、c1-c6烷基双取代胺基、c1-c6烷氧基、c1-c6烷基羰氧基、c1-c6环烷基羰氧基、杂环基羰氧基、c1-c6烷氧基羰基、c1-c6环烷氧基羰基、杂环氧基羰基、c1-c6烷基羰胺基、c1-c6环烷基羰胺基、杂环基羰胺基、胺基羰基、c1-c6烷氧甲酰胺基、c1-c6烷巯基、羟基烷氧基、糖残基、磺酸基、磷酸基、多羟基c1-c6烷氧基羰基、羧基c1-c6烷氧基、羧基c1-c6烷基甲酰氧基中的一个或多个。
[0019]
再进一步优选地,r选自h、取代或未取代的c1-c8烷基、取代或未取代的c2-c8烯基。
[0020]
再进一步优选地,r选自h、取代或未取代的c1-c8烷基。
[0021]
再进一步优选地,r选自h、c1-c8烷基。
[0022]
再进一步优选地,r选自h、c1-c5烷基。
[0023]
再进一步优选地,r选自h、c1-c3烷基。
[0024]
再进一步优选地,r选自h、甲基。
[0025]
优选地,所述制备方法包括以下步骤:
[0026]
(1)将对甲氧基苯肼盐酸盐经1,4-丁二醛或者2,5-己二酮保护得到化合物1;
[0027]
(2)将步骤(1)得到的化合物1和对氯苯甲酰氯发生酰化反应得到化合物2;
[0028]
(3)将步骤(2)得到的化合物2用水合肼脱保护得到化合物3;
[0029]
(4)将步骤(3)得到的化合物3在酸作用下和乙酰丙酸发生关环反应得到吲哚美辛。
[0030]
进一步优选地,步骤(1)具体为:溶剂溶解对甲氧基苯肼盐酸盐,0-15℃加入1,4-丁二醛或者2,5-己二酮,0-15℃反应8-15h,经后处理得化合物1。
[0031]
更进一步优选地,所述溶剂选自甲醇、乙醇、异丙醇、二氯甲烷中的至少一种。
[0032]
更进一步优选地,所述对甲氧基苯肼盐酸盐与1,4-丁二醛或者2,5-己二酮的摩尔比为1:1.1-2.0。
[0033]
更进一步优选地,所述加入1,4-丁二醛或者2,5-己二酮和所述反应的温度为为0-10℃;所述反应的时间为10h。
[0034]
更进一步优选地,所述后处理的步骤包括:控温0-5℃滴加饱和碳酸氢钠溶液,升
温至20-30℃,有机相减压浓缩,加入正庚烷,搅拌,抽滤,得到化合物1。
[0035]
进一步优选地,步骤(2)具体为:溶剂溶解化合物1,加入有机碱,10-30℃加入4-氯苯甲酰氯,10-30℃保温反应3-8h,后处理得化合物2。
[0036]
更进一步优选地,所述化合物1与有机碱摩尔比为1:1.2-3.0、化合物1与对氯苯甲酰氯摩尔比为1:1.0-1.5。
[0037]
更进一步优选地,所述溶剂选自二氯甲烷、氯仿、氯苯、二氯苯中的至少一种。
[0038]
更进一步优选地,所述有机碱选自三乙胺、咪唑、吡啶、哌啶中的至少一种,更进一步优选为三乙胺和吡啶的混合物,更进一步地,三乙胺和吡啶的摩尔比为30:1。
[0039]
更进一步优选地,所述加入4-氯苯甲酰氯和所述保温反应的温度为10-20℃;所述保温反应的时间为5h。
[0040]
更进一步优选地,所述后处理步骤包括:控温0-20℃加水淬灭反应、搅拌,再经分液、洗涤、减压浓缩、结晶。
[0041]
进一步优选地,步骤(3)具体为:溶剂溶解化合物2,加入水合肼,60-90℃反应40-60h,后处理得化合物3。
[0042]
更进一步优选地,所述溶剂选自甲醇、乙醇、异丙醇中的至少一种。
[0043]
更进一步优选地,所述化合物2与水合肼的摩尔比为1:5-15,最优选为1:10。
[0044]
更进一步优选地,所述反应为80℃反应48h。
[0045]
更进一步优选地,所述后处理步骤包括:5-10℃将反应液加入到水中,搅拌,抽滤,洗涤,将所得滤饼用水分液,收集有机相后重结晶。
[0046]
进一步优选地,步骤(4)具体为:溶剂溶解化合物3,加入酸和乙酰丙酮酸,100-120℃反应40-60h,后处理得吲哚美辛。
[0047]
更进一步优选地,所述溶剂选自甲苯、二甲苯、氯苯中的至少一种。
[0048]
更进一步优选地,所述酸选自甲磺酸和/或对甲苯磺酸。
[0049]
更进一步优选地,所述化合物3与酸的摩尔比为1:0.1-1.0,化合物3与乙酰丙酮酸摩尔比为1:1.2-2.0。
[0050]
更进一步优选地,所述后处理步骤包括:降温至20-30℃,将反应液加入到饱和碳酸氢钠中,搅拌,分液,收集水相,水相萃取,调节ph至2-3,20-30℃搅拌,过滤,滤饼重结晶。
[0051]
本发明的有益效果为:
[0052]
(1)原料廉价易得,收率和纯度均较高,中间体稳定质量可控,可以满足gmp的生产要求;(2)工艺操作相对简单,适合规模化工业生产。
附图说明
[0053]
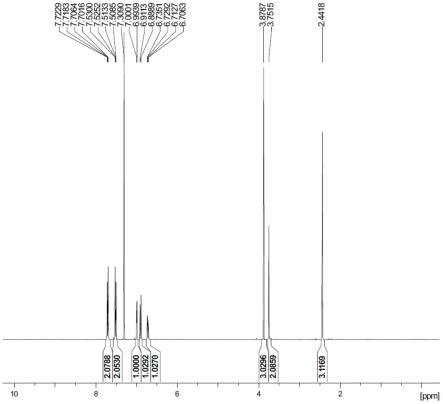
图1为实施例1制备的吲哚美辛的氢谱图。
具体实施方式
[0054]
以下非限制性实施例可以使本领域的普通技术人员更全面的理解本发明,但不以任何方式限制本发明。下述内容仅仅是对本技术要求保护的范围的示例性说明,本领域技术人员可以根据所公开的内容对本技术的发明作出多种改变和修饰,而其也应当属于本技术要求保护的范围之中。
[0055]
下面以具体实施例的方式对本发明作进一步的说明。本发明实施例中所使用的各种化学试剂如无特殊说明均通过常规商业途径获得。若无特殊说明,下文中所述含量均为质量含量。
[0056]
下述实施例中,对甲氧基苯肼盐酸盐购自长沙俊宇医药科技有限公司,货号为jymp-20220116。
[0057]
实施例1
[0058]
(1)于5l三口瓶中加入对甲氧基苯肼盐酸盐(100g,0.57mol),加入二氯甲烷(1l),控温0-10℃加入2,5-己二酮(71.5g,0.63mol),加完后0-10℃反应10h。控温0-5℃滴加饱和碳酸氢钠溶液(0.5l),升温至20-30℃,分液,收集下层有机相。有机相减压浓缩至约1/3体积,20-30℃滴加正庚烷(2l)(滴加时间约1h)。20-30℃搅拌0.5h,抽滤,得到n-(4-甲氧基苯基)-2,5-二甲基-1h-吡咯-1-胺(化合物1)105g,收率85%,纯度99.5%。
[0059]
(2)于5l三口瓶中加入步骤(1)得到的n-(4-甲氧基苯基)-2,5-二甲基-1h-吡咯-1-胺1(100.0g,0.46mol),加入二氯甲烷(1l)、三乙胺(112g,0.69mol)、4-n,n-二甲基吡啶(102.0g,23mmol),控温10-20℃滴加4-氯苯甲酰氯(65.5g,0.64mol),加完后10-20℃保温反应5h。控温10-20℃滴加水(0.5l),搅拌0.5h。分液,用饱和氯化钠溶液(0.5l)洗有机相4次,用硫酸钠干燥,有机相减压浓缩至约1/4体积。20-30℃滴加正庚烷(4l)(滴加时间约1h)。20-30℃搅拌0.5h,抽滤,得化合物2 130g,收率80%,纯度99.4%。
[0060]
(3)于5l三口瓶中加入步骤(2)中所得化合物2(120g,0.34mol),加入乙醇(600ml)、80%水合肼(213g,3.4mol),80℃下反应48h。控温5-10℃将反应液加入到水中(1.2l),搅拌0.5h。抽滤,滤饼用水(1.2l*2)淋洗两次。上述湿品溶解于二氯甲烷(360ml),用水(200ml*2)分液,收集上层有机相后加入正庚烷(720ml)重结晶得到化合物3 81.6g,87%收率,纯度99.8%。
[0061]
(4)于5l三口瓶中加入步骤(3)中所得化合物3(80g,0.29mol),加入甲苯(600ml)、三氟乙酸(33g,0.29mol)、乙酰丙酮酸(67g,0.58mol),110℃下反应48h。降温至20-30℃,将反应液加入到饱和碳酸氢钠中(600ml),搅拌0.5h。分液,收集水相,水相用甲苯(300ml)萃取一次;水相用浓盐酸调节ph至2-3,20-30℃搅拌0.5h,过滤,收集滤饼。滤饼用乙酸乙酯(170ml)和正庚烷(330ml)重结晶得到产品67g,65%收率,纯度99.90%。
[0062]
所得产物结构为:
[0063][0064]
氢谱数据:1h-nmr(400mhz,cdcl3):δ2.44(s,3h),3.75(s,2h),3.88(s,3h),3.31(m,1h)6.72(dd,j=2.4hz,7.2hz,1h),6.89(d,j=7.2hz,1h),6.99(d,j=2.4hz,1h),7.35(d,j=8.0hz,1h),7.52(dd,j=7.2hz,2.4hz,2h),7.71(dd,j=7.2hz,2.4hz,2h)。
[0065]
实施例2
[0066]
与实施例1不同的是,步骤(4)中将三氟乙酸换成对甲苯磺酸(0.1eq),其余皆相
同。反应最终得到产品72g,70%收率,纯度99.90%。
[0067]
氢谱数据:1h-nmr(400mhz,cdcl3):δ2.44(s,3h),3.75(s,2h),3.88(s,3h),3.31(m,1h)6.72(dd,j=2.4hz,7.2hz,1h),6.89(d,j=7.2hz,1h),6.99(d,j=2.4hz,1h),7.35(d,j=8.0hz,1h),7.52(dd,j=7.2hz,2.4hz,2h),7.71(dd,j=7.2hz,2.4hz,2h)。
[0068]
实施例3
[0069]
(1)于5l三口瓶中加入对甲氧基苯肼盐酸盐(100g,0.57mol),加入二氯甲烷(1l),控温0-10℃加入1,4-丁二醛(54g,0.63mol),加完后0-10℃反应10h。控温0-5℃滴加饱和碳酸氢钠溶液(0.5l),升温至20-30℃,分液,收集下层有机相。有机相减压浓缩至约1/3体积,20-30℃滴加正庚烷(2l)(滴加时间约1h)。20-30℃搅拌0.5h,抽滤,得到n-(4-甲氧基苯基)-1h-吡咯-1-胺86g,收率80%,纯度99.2%。
[0070]
(2)于5l三口瓶中加入步骤(1)得到的n-(4-甲氧基苯基)-1h-吡咯-1-胺1(86.0g,0.46mol),加入二氯甲烷(1l)、三乙胺(112g,0.69mol)、4-n,n-二甲基吡啶(102.0g,23mmol),控温10-20℃滴加4-氯苯甲酰氯(65.5g,0.64mol),加完后10-20℃保温反应5h。控温10-20℃滴加水(0.5l),搅拌0.5h。分液,用饱和氯化钠溶液(0.5l)洗有机相4次,用硫酸钠干燥,有机相减压浓缩至约1/4体积。20-30℃滴加正庚烷(4l)(滴加时间约1h)。20-30℃搅拌0.5h,抽滤,得化合物2 124g,收率83%,纯度99.4%。
[0071]
(3)于5l三口瓶中加入步骤(2)中得到的苯甲酰化合物2(120g,0.38mol),加入乙醇(600ml)、80%水合肼(238g,3.8mol),80℃下反应48h。控温5-10℃将反应液加入到水中(1.2l),搅拌0.5h。抽滤,滤饼用水(1.2l*2)淋洗两次。上述湿品溶解于二氯甲烷(360ml),用水(200ml*2)分液,收集上层有机相后加入正庚烷(720ml)重结晶得到化合物3 89g,85%收率,纯度99.8%。
[0072]
(4)于5l三口瓶中加入步骤(3)中所得化合物3(80g,0.29mol),加入甲苯(600ml)、三氟乙酸(33g,0.29mol)、乙酰丙酮酸(67g,0.58mol),110℃下反应48h。降温至20-30℃,将反应液加入到饱和碳酸氢钠中(600ml),搅拌0.5h。分液,收集水相,水相用甲苯(300ml)萃取一次;水相用浓盐酸调节ph至2-3,20-30℃搅拌0.5h,过滤,收集滤饼。滤饼用乙酸乙酯(170ml)和正庚烷(330ml)重结晶得到产品70g,68%收率,纯度99.90%。
[0073]
所得产物结构为:
[0074][0075]
氢谱数据:1h-nmr(400mhz,cdcl3):δ2.44(s,3h),3.75(s,2h),3.88(s,3h),3.31(m,1h)6.72(dd,j=2.4hz,7.2hz,1h),6.89(d,j=7.2hz,1h),6.99(d,j=2.4hz,1h),7.35(d,j=8.0hz,1h),7.52(dd,j=7.2hz,2.4hz,2h),7.71(dd,j=7.2hz,2.4hz,2h)。
[0076]
实施例4
[0077]
与实施例1不同的是,步骤(4)中将三氟乙酸换成甲磺酸,其余皆相同。反应最终得到产品61g,收率59%,纯度99.8%。
[0078]
氢谱数据:1h-nmr(400mhz,cdcl3):δ2.44(s,3h),3.75(s,2h),3.88(s,3h),3.31(m,1h)6.72(dd,j=2.4hz,7.2hz,1h),6.89(d,j=7.2hz,1h),6.99(d,j=2.4hz,1h),7.35(d,j=8.0hz,1h),7.52(dd,j=7.2hz,2.4hz,2h),7.71(dd,j=7.2hz,2.4hz,2h)。
[0079]
实施例5
[0080]
与实施例1不同的是,步骤(2)中将三乙胺换成咪唑,其余皆相同。
[0081]
咪唑结构:
[0082]
反应最终得到化合物2 125g,收率77%,纯度99.2%。
[0083]
氢谱数据:1h-nmr(400mhz,cdcl3):δ2.44(s,3h),3.75(s,2h),3.88(s,3h),3.31(m,1h)6.72(dd,j=2.4hz,7.2hz,1h),6.89(d,j=7.2hz,1h),6.99(d,j=2.4hz,1h),7.35(d,j=8.0hz,1h),7.52(dd,j=7.2hz,2.4hz,2h),7.71(dd,j=7.2hz,2.4hz,2h)。
[0084]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
