1.本发明属于有机化工中间体合成与分离技术领域,具体而言,涉及酸碱协同催化甲醛与丙醛连续缩合制备甲基丙烯醛的方法。
背景技术:
2.甲基丙烯醛是一种有着重要用途的有机合成中间体,可以用来合成甲基烯丙醇、甲基丙烯酸和甲基丙烯酸酯等化工原料。其中,甲基烯丙醇可以用来合成甲基烯丙基聚氧乙烯醚(hpeg),一种非常重要的聚羧酸类减水剂,其减水效率高、耐久性长、增强效果好、对环境友好,已大规模应用于高速铁路、高速公路、跨海大桥、海底隧道、高层建筑等大型工程;甲基丙烯酸(maa)是重要的有机化工原料和聚合单体,可用于制造树脂、塑料、涂料、黏合剂、润滑剂、木材和软木的浸润剂、纸张上光剂等;品种多样的甲基丙烯酸特种酯附加值高,经济效益好,在涂料、粘接剂、树脂改性剂、交联剂、合成橡胶、隐形眼镜等领域应用广泛;甲基丙烯酸甲酯(mma)主要用作pmma树脂和pvc抗冲击改性剂mbs的合成单体,其中:pmma透光性、耐候性和光泽度好,在液晶显示屏、防弹玻璃和光导纤维等领域广泛应用;mbs主要用于纺织上浆剂、金属粘结剂等。此外,甲基丙烯醛还可用于合成异丁醛,异丁醛用来制造抗氧化剂、增塑剂、果子精油和合成药物等;甲基丙烯醛还是高档香精香料兔耳草醛和铃兰醛等的合成原料。因甲基丙烯醛下游产品如有机玻璃、聚羧酸减水剂、香精香料等的应用日益普及,未来我国年均需求量将达到百万吨级,因此开发低成本的甲基丙烯醛连续清洁合成技术,具有很好的社会效益和经济效益。
3.现有甲基丙烯醛合成方法主要有异丁烯(叔丁醇)氧化法、异丁烷氧化法、甲醛和丙醛缩合法。异丁烯(叔丁醇)氧化法工艺较成熟,主要缺点是装置投资大、反应温度高、产物甲基丙烯醛选择性较低;异丁烷氧化法原料廉价易得,但由于异丁烷性质稳定,反应条件苛刻,原料异丁烷有效利用率和产物甲基丙烯醛选择性很低;而甲醛和丙醛缩合法,原料来源广泛,价格低廉,反应条件温和,设备简单,装置投资低,预期采用该路线合成甲基丙烯醛具有较强的竞争优势。
4.目前,工业上煤制甲醇、甲醇制甲醛、甲醇制乙烯、乙烯氢甲酰化制丙醛等技术的成功应用,为煤基路线制备甲基丙烯醛奠定了基础。目前工业上生产甲基丙烯醛主要采用基于石油资源的异丁烯氧化法,甲基丙烯醛选择性较低,而且国内石油资源短缺,煤炭储量丰富。因此,开发煤基路线的甲醛与丙醛缩合生产甲基丙烯醛技术以及自主知识产权的清洁生产新工艺,不仅符合我国国情,且具有很好的经济性和现实意义。
5.从原料甲醛和丙醛出发制备甲基丙烯醛有两种方法:一种是采用强碱性催化剂如koh、醇盐等,由甲醛和丙醛经羟醛缩合(aldol缩合)反应合成甲基丙烯醛,该方法因原料丙醛自身缩合生成大量副产物2-甲基-2-戊烯醛,导致原料有效利用率和甲基丙烯醛选择性较低,进而提高了原料成本;此外,强碱催化剂用量大,且无法循环而一次性使用,导致污染严重,对环境不友好。另一种是采用有机胺-有机酸复合催化剂,由甲醛和丙醛经mannich缩合反应合成甲基丙烯醛,这种方法原料转化率和利用率高,甲基丙烯醛选择性好,生产成本
低,催化剂用量少,而且采用适宜的工艺还可以循环套用,环境污染小,属于安全清洁工艺。
6.在现有报道的甲醛和丙醛mannich缩合制备甲基丙烯醛的方法中,大都采用间歇反应工艺。例如:cn104003856公开了一种以甲醛和丙醛为原料制备甲基丙烯醛的方法,采用仲醇胺和有机酸混合催化剂,原料摩尔比为甲醛:丙醛:醇胺=0.5~1.5:1.0:1.0~10.0,催化剂体系中各物质摩尔比为醇胺:醌类物质:有机酸=1.0:0.0001~0.01:0.5~1.5,反应温度10~60℃,反应时间60min,丙醛转化率超过97%,选择性99%,但催化剂最小用量都超过30wt%。cn102659542报道了一种以离子液体催化甲醛与丙醛缩合制备甲基丙烯醛的方法,在温度0~90℃、物料摩尔比甲醛:丙醛:离子液体=0.8~1.2:1.0:0.5~1.0和体系水含量1~60%下反应0.1~180min制备得到甲基丙烯醛,该方法反应条件温和,转化率和选择性较高,但离子液体催化剂用量太大(占原料重量50%以上),而且离子液体与反应产物难分离。cn104557490使用多聚甲醛解聚替代甲醛水溶液,采用吗啉盐酸盐催化剂,控制反应温度50~60℃、反应物重量比多聚甲醛:丙醛:吗啉盐酸盐为0.5:1.0:0.1下,甲基丙烯醛收率97%,选择性接近100%;该方法可以提高甲醛浓度,减少采用甲醛水溶液原料带入的反应废水量,但盐酸的挥发性和强酸性对生产设备有更高的要求,而且催化剂用量超过6.0wt%。cn104311403使用多聚甲醛在有机溶剂中解聚所得的甲醛有机溶液替代甲醛水溶液,在温度10~90℃、物料摩尔比丙醛:甲醛:有机溶剂:催化剂中有机胺=1.0:1.0~2.0:0.3~5.0:0.2~2.0、体系水含量2~40wt%下反应0.1~3h,原料转化率及产物选择性均在98%以上,该方法从源头上减少了水量,与用商品37%甲醛溶液做原料相比,大幅降低了废水量,但因使用有机溶剂,不仅使产物分离变得更加复杂,引入了新的溶剂污染源,而且提高了生产成本。上述方法对催化剂种类和甲醛的形态进行了调整,但均采用间歇装置在常压和较低反应温度下进行缩合反应,催化剂用量普遍很高且不能循环套用,导致环境污染严重和生产成本高。
7.针对间歇工艺催化剂低效的问题,一些研究者探索了微通道反应技术,拟提高催化反应效率。例如,us4496770报道的合成甲基丙烯醛的方法中,采用仲胺(二甲胺、二乙胺、二丁胺、哌嗪等)和乙酸作为催化剂,在摩尔比甲醛:丙醛:乙酸:仲胺=1.0:1.0:0.003~0.005:0.003、温度161~186℃、压力48~50bar、物料接触时间6.9s下,得到油相中mal收率为92~98%。专利cn107074715也对微通道反应技术进行了报道,以乙酸和二甲胺为催化剂,在摩尔比甲醛:丙醛:乙酸:二甲胺=1.0:1.0:0.053~0.056:0.058~0.061、温度160℃、压力55bar和接触时间9.4~9.7s下反应,mal收率高达97%。上述方法采用微通道反应装置,在升高温度的同时提高了反应体系压力,反应速率明显加快,并且温度升高使mannich缩合反应与mannich碱分解反应同时进行,大大降低了催化剂用量。但是,微通道反应技术也存在一定的局限性,比如反应条件极为苛刻,温度超过160℃和压力超过48bar,而且微通道反应装置造价高,不适用于大规模工业化生产等。另外,微通道反应器因微管过小,对于有聚合倾向的热敏性物料体系,易堵塞而难以实现长周期运行,更难以实现含催化剂和聚合物或焦油的物料循环。微通道反应器对于强放热、快反应、易流动的均相体系具有优异的反应效果和良好的安全性,但对于存在固体或粘稠物料或有聚合倾向的热敏性物料体系,因存在堵塞问题而无法满足长时间运行的生产要求。
8.为了解决微通道反应技术生产规模小、易堵塞、物料难循环的问题,有研究者探究了连续管式反应技术。如cn113248359报道了一种在管式反应器中制备甲基丙烯醛的方法,
在摩尔比甲醛:丙醛:有机胺:有机酸=1.05:1.0:1.0~1.2:1.0~1.2、温度50~70℃、停留时间1~2小时的条件下进行缩合反应,反应产物直接送入精馏塔进行催化剂解析和分离,在常压、塔釜温度100~130℃下连续精馏,塔顶得到产物甲基丙烯醛和部分水,冷凝后在分相器中得到上层产品甲基丙烯醛,以产品计丙醛转化率最高为99%,mal收率最高为98%。该方法因管式反应温度低,添加的催化剂量太大,在原料体系中催化剂占比达到50wt%以上,并且精馏塔塔釜温度较高,解析出来的催化剂会使产物甲基丙烯醛自身缩合加剧,进而严重影响精馏收率。cn110882702公开了一种酸碱复合催化剂以及使用其制备甲基丙烯醛的方法,采用釜式反应器与管式反应器相组合的方式,甲醛与丙醛摩尔比为1.0:0.1~10.0、催化剂用量为反应物重量的0.01~4倍,先在釜式反应器中50~70℃下反应15~40min;然后在管式反应器中进行,反应温度110~150℃,压力0~5mpa,停留时间3秒~60分钟,原料转化率及产物选择性均在93%以上。该方法没有充分发挥管式反应器的优势,催化剂用量并没有得到降低,甚至采用的复合催化剂用量更大,其效果并不比有机酸-有机胺组合催化剂有优势,而且釜式反应与管式反应组合使反应系统更复杂,增加了反应装置投资。
9.为了解决上述专利中存在的催化剂用量大、三废多、生产规模小等问题,非常有必要设计开发一种新的生产甲基丙烯醛的方法。
技术实现要素:
10.为了解决上述问题,本发明提供一种采用管式反应工艺的甲基丙烯醛连续、稳定和高效生产方法,该方法催化剂效率高、用量少且可循环使用,生产过程物耗能耗低且安全环保。
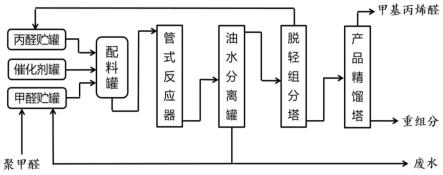
11.本发明的技术方案如下:一种甲醛和丙醛缩合连续生产甲基丙烯醛的方法,包括如下步骤:
12.(1)将甲醛原料、丙醛、催化剂和阻聚剂在常温下搅拌均匀后,连续送入管式反应器中,进行缩合反应得到缩合反应产物,所述催化剂为有机胺-有机酸组合催化剂;
13.(2)将步骤(1)得到的所述缩合反应产物进行油水分离,得到醛类油相物料和含催化剂、阻聚剂、甲醇和醛的水相物料;
14.(3)向步骤(2)中分离出的所述油相物料中加入适量阻聚剂后,送入脱轻组分塔精馏,塔顶采出丙醛物料循环用于步骤(1)的反应原料,塔釜采出粗甲基丙烯醛物料;
15.(4)将步骤(3)得到的所述粗甲基丙烯醛物料送入产品精馏塔,塔顶采出甲基丙烯醛产品,塔底重组分作为副产品回收;
16.(5)将步骤(2)中分离出的所述水相物料,部分循环并用于加入聚甲醛进行解聚配制甲醛原料,其余部分作为废水进行处理。
17.本发明所述方法的步骤(5)中,将分离出的水相物料分为两部分进行处理,一部分通过加入聚甲醛解聚成甲醛水溶液,保持原料甲醛水溶液浓度和物料总量不变而被重复利用;剩余部分因甲醛与丙醛缩合反应也会生成水,因此为保持反应体系连续稳定运行需外排。
18.进一步的,步骤(1)中,所述甲醛原料为浓度30~55%的甲醛溶液;所述有机胺-有机酸组合催化剂中,所述有机胺选自伯胺、仲胺、叔胺、多胺、咪唑、吡啶、季铵盐、醇胺、氨基酸或酰胺中的一种或多种,所述有机酸选自羧酸、磺酸或膦酸中的一种或多种。
19.本发明中,所述甲醛溶液可通过水吸收纯甲醛制得,或通过甲缩醛、三聚甲醛或多聚甲醛中的至少一种在水中解聚得到,或通过福尔马林溶液配制得到。甲醛溶液的浓度低于30%,会造成反应后废水量过大,且生产中能耗较大;而浓度高于55%的甲醛溶液不稳定,易生产聚甲醛,因此本发明中优选30~55%浓度的甲醛溶液。
20.进一步的,所述缩合反应的甲醛原料为35~52%的甲醛溶液;所述有机胺选自氨水、甲胺、乙胺、丙胺、丁胺、苄胺、乙醇胺、异丙醇胺、乙二胺、丙二胺、戊二胺、己二胺、二乙烯三胺、三乙烯四胺、二甲胺、二乙胺、二丙胺、二异丙胺、二丁胺、二异丁胺、二环己胺、二苯胺、二苄胺、四氢吡咯、哌啶、哌嗪、吗啉、吡啶、吡咯、二乙醇胺、二异丙醇胺、三甲胺、三乙胺、三丙胺、三乙烯二胺、六亚甲基四胺、氨基乙酸、氨基丙酸、氨基丁酸、缬氨酸、赖氨酸、苯丙氨酸、谷氨酸、丁内酰胺、戊内酰胺或己内酰胺中的一种或多种;
21.所述有机酸选自甲酸、乙酸、丙酸、正丁酸、异丁酸、正戊酸、特戊酸、苯甲酸、环烷酸、一氯乙酸、二氯乙酸、三氯乙酸、甲磺酸、对甲苯磺酸、三氟甲磺酸、乙二酸、丙二酸、丁二酸、戊二酸、己二酸、庚二酸、癸二酸、对苯二甲酸、邻苯二甲酸或间苯二甲酸中的一种或多种。
22.优选的,步骤(1)中,所述甲醛原料选自37%、44%或50%浓度的工业甲醛溶液中的至少一种;所述有机胺选自乙胺、丙胺、乙二胺、1,3-丙二胺、1,6-己二胺、二甲胺、二乙胺、二丙胺、二丁胺、四氢吡咯、哌啶、哌嗪、二乙醇胺、三乙胺、甘氨酸、赖氨酸、3-氨基丙酸、吡咯烷酮或哌啶酮中的一种或多种;
23.所述有机酸选自甲酸、乙酸、丙酸、正丁酸、异丁酸、苯甲酸、三氯乙酸、对甲苯磺酸、乙二酸、丙二酸、丁二酸、戊二酸或己二酸中的一种或多种。
24.进一步的,步骤(1)中,在所述管式反应器内,所述缩合反应的物料摩尔配比为丙醛:甲醛:有机胺:有机酸=1.000:(0.800~1.300):(0.0001~0.080):(0.0001~0.080);所述缩合反应条件为温度60~160℃、压力0.1~5.0mpa、物料停留时间10~180min。
25.进一步的,所述缩合反应的物料摩尔配比为丙醛:甲醛:有机胺:有机酸=1.000:(0.900~1.150):(0.0005~0.050):(0.0005~0.050);所述缩合反应条件为温度80~145℃、压力0.3~3.0mpa、物料停留时间20~90min。
26.更进一步的,所述缩合反应的物料摩尔配比为丙醛:甲醛:有机胺:有机酸=1.000:(1.000~1.100):(0.001~0.020):(0.001~0.020);所述缩合反应条件为温度100~130℃、压力0.6~1.5mpa、物料停留时间30~60min。
27.进一步的,步骤(2)中,所述缩合反应产物在油水分离罐中通过静置萃取进行油水分离,上层为油相物料,下层为水相物料,静置萃取时间0.5~24h、温度5~55℃。
28.优选的,静置萃取时间1~10h、温度15~50℃。
29.更优选的,静置萃取时间2~8h、温度20~40℃。
30.进一步的,所述脱轻组分塔的操作条件为压力20~100kpa、塔釜温度25~70℃、塔顶温度15~60℃和回流比30~60:1;所述产品精馏塔的操作条件为压力10~100kpa、塔釜温度35~105℃、塔顶温度15~70℃和回流比1:6~30。
31.进一步的,所述脱轻组分塔的操作条件为压力30~90kpa、塔釜温度30~65℃、塔顶温度20~55℃和回流比35~55:1;所述产品精馏塔的操作条件为压力20~85kpa、塔釜温度45~90℃、塔顶温度25~65℃和回流比1:8~25。
32.更进一步的,所述脱轻组分塔的操作条件为压力45~75kpa、塔釜温度45~55℃、塔顶温度30~50℃和回流比40~50:1;所述产品精馏塔的操作条件为压力30~70kpa、塔釜温度60~80℃、塔顶温度35~55℃和回流比1:10~20。
33.进一步的,所述阻聚剂选自酚、醌、氮氧自由基或硫氮杂蒽中的一种或任意组合。
34.进一步的,所述阻聚剂选自对苯二酚、对羟基苯甲醚、对甲酚、对叔丁基酚、对叔丁基邻苯二酚(tbc)、2-叔丁基对苯二酚、2,6-二叔丁基对甲苯酚(bht)、2,5-二叔丁基对苯二酚、对苯醌、4-羟基-2,2,6,6-四甲基哌啶氮氧自由基(zj-701)、三(1-羟基-2,2,6,6-四甲基哌啶-4-)亚磷酸酯(zj-705)或吩噻嗪中的一种或任意组合。
35.优选的,所述阻聚剂选自对苯二酚、对羟基苯甲醚、对苯醌、zj-701、zj-705或吩噻嗪中的一种或任意组合。
36.进一步的,步骤(1)中,所述阻聚剂的加入量为甲醛原料、丙醛和催化剂总质量的20~2000ppm;步骤(3)中,所述阻聚剂的加入量为所述油相物料质量的20~2000ppm。
37.进一步的,步骤(1)中,所述阻聚剂的加入量为甲醛原料、丙醛和催化剂总质量的50~1000ppm;步骤(3)中,所述阻聚剂的加入量为所述油相物料质量的50~1000ppm。
38.优选的,步骤(1)中,所述阻聚剂的加入量为甲醛原料、丙醛和催化剂总质量的100~500ppm;步骤(3)中,所述阻聚剂的加入量为所述油相物料质量的100~500ppm。
39.本发明具有以下有益效果:
40.(1)与现有工业生产采用等温列管固定床反应器的异丁烯氧化法相比,原料甲醛和丙醛价廉易得,缩合反应条件温和,为液态均相反应,反应产物甲基丙烯醛选择性高;反应及分离工艺简单,装置投资低;能耗物耗低,生产成本低。
41.(2)与现有同类技术采用釜式反应器的甲醛-丙醛间歇缩合法相比,在加压和较高温度下进行液态均相连续缩合反应,催化反应效率高,原料转化率和产物甲基丙烯醛选择性高;且大幅降低了催化剂用量,过程更环保,生产成本更低。
42.(3)与现有同类技术采用微通道反应器的甲醛-丙醛连续缩合法相比,反应装置造价低,生产规模大,反应条件温和,反应温度和压力低,反应物料尤其是过程中生成的微量聚合物或焦油不堵塞反应器,装置运行周期长,反应平稳。
43.(4)与现有同类专利技术相比,本发明采用管式反应器、高效分离工艺、阻聚技术和新型高效的有机酸-有机胺组合催化剂,生产甲基丙烯醛的过程连续、稳定、高效,装置产能大、操作简便,可大规模生产;未反应完全的丙醛和游离态甲醛可100%循环使用,而且60~75%的催化剂、阻聚剂和工艺水可循环使用,原料甲醛、丙醛催化剂和阻聚剂利用率高,操作条件温和,产生的三废少,过程安全环保;产品甲基丙烯醛收率高、品质好、成本低。
附图说明
44.图1为本发明甲醛与丙醛缩合连续生产甲基丙烯醛的反应和分离精制工艺流程示意图。
具体实施方式
45.下面用具体实施例来对本发明的技术方案进行清楚、完整地描述,但所列举的实施例仅仅是本发明的部分实施例,而不是全部。基于本发明中的实施例,本领域技术人员在
没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
46.需要说明的是,实施例中fa、pl和mal分别代表甲醛、丙醛和甲基丙烯醛,c、s、u、y、ni和mi分别表示转化率、选择性、有效利用率、分离精制收率、i物质的摩尔数和i物质的质量。则丙醛转化率c
pl
、甲基丙烯醛选择性s
mal
、甲醛有效利用率u
fa
和甲基丙烯醛的分离精制收率计算公式为:
47.c
pl
=(反应进料中n
pl-反应产物中n
pl
)/反应进料中n
pl
×
100%;
48.s
mal
=反应产物中n
mal
/(反应进料中n
pl-反应产物中n
pl
)
×
100%;
49.u
fa
=反应产物中n
mal
/反应进料中n
fa
×
100%;
50.y
mal
=分离精制终产品m
mal
/分离精制进料中m
mal
×
100%。
51.实施例1~2和对比例1~2为不同反应方式和催化剂用量的缩合反应效果
52.实施例1
53.采用37%的工业甲醛溶液(含约10%甲醇)为原料。将1.050mol甲醛、1.000mol丙醛、0.006mol乙酸、0.006mol二乙胺和100ppm对苯二酚分别加入三口烧瓶中,搅拌均匀后,用平流泵连续送入外径6mm、内径4mm和长400mm的管式反应器中,在温度110℃、压力0.8mpa和停留时间45min下进行甲醛与丙醛的mannich缩合反应。收集反应液进行气相色谱分析,并根据分析结果计算得到:丙醛转化率94.01%、甲基丙烯醛选择性96.54%和甲醛有效利用率86.43%。
54.实施例2
55.试验过程和原料用量均同实施例1,仅将催化剂和阻聚剂的用量改变,乙酸、二乙胺和对苯二酚用量分别提高到0.010mol、0.010mol和200ppm。丙醛转化率96.58%、甲基丙烯醛选择性98.67%和甲醛有效利用率90.76%。
56.对比例1
57.采用37%的工业甲醛溶液(含约10%甲醇)为原料。将1.050mol甲醛、1.000mol丙醛、1.000mol乙酸、1.000mol二乙胺和250ppm对苯二酚分别加入三口烧瓶中,在温度45℃、常压下搅拌,进行甲醛与丙醛的mannich缩合反应45min。对烧瓶中的反应液进行气相色谱分析,并根据分析结果计算得到:丙醛转化率99.25%、甲基丙烯醛选择性91.45%和甲醛有效利用率86.44%。
58.对比例2
59.试验过程和原料用量均同对比例1,仅将催化剂和阻聚剂的用量改变,乙酸、二乙胺和对苯二酚用量分别降低到0.100mol、0.100mol和100ppm。丙醛转化率52.13%、甲基丙烯醛选择性88.29%和甲醛有效利用率43.83%。
60.从实施例1~2及对比例1~2可以看出,采用连续管式反应器,较采用间歇釜式反应器催化剂用量大大降低,而且产物甲基丙烯醛选择性更高。
61.实施例3和对比例3~4为不同有机胺属性催化剂的缩合反应效果
62.实施例3
63.采用三聚甲醛解聚配制50%的甲醛溶液为原料。将1.000mol甲醛、1.000mol丙醛、0.015mol乙酸、0.010mol二丙胺和200ppm对羟基苯甲醚分别加入三口烧瓶中,搅拌均匀后,用平流泵连续送入外径6mm、内径4mm和长400mm的管式反应器中,在温度105℃、压力0.6mpa和停留时间60min下进行甲醛与丙醛的mannich缩合反应。收集反应液进行气相色谱分析,
并根据分析结果计算得到:丙醛转化率94.68%、甲基丙烯醛选择性98.59%和甲醛有效利用率93.34%。
64.对比例3
65.试验过程和各物质用量均同实施例3,仅将二丙胺替换为丙胺。丙醛转化率16.66%、甲基丙烯醛选择性98.39%和甲醛有效利用率16.39%。
66.对比例4
67.试验过程和各物质用量均同实施例3,仅将二丙胺替换为三丙胺。丙醛转化率24.05%,甲基丙烯醛选择性98.02%和甲醛有效利用率23.57%。
68.从实施例3和对比例3-4可知,对于不同属性的有机胺,其对本发明的聚合反应有不同的催化效果,其中,仲胺的催化性能最佳,伯胺和叔胺催化活性较低。
69.实施例4~10和对比例5为不同组合催化剂和阻聚剂的缩合反应效果
70.实施例4
71.采用三聚甲醛解聚配制50%的甲醛溶液为原料。将1.000mol甲醛、1.000mol丙醛、0.008mol对甲苯磺酸(ptsa)、0.010mol二乙胺和150ppm对苯醌分别加入三口烧瓶中,搅拌均匀后,用平流泵连续泵入外径6mm、内径4mm和长400mm的管式反应器中,在温度100℃、压力0.7mpa和停留时间60min下进行甲醛与丙醛的mannich缩合反应。收集反应液进行气相色谱分析,并根据分析结果计算得到:丙醛转化率94.32%、甲基丙烯醛选择性97.88%和甲醛有效利用率92.32%。
72.实施例5
73.采用三聚甲醛解聚配制50%的甲醛溶液为原料。将1.000mol甲醛、1.000mol丙醛、0.010mol乙酸、0.008mol四氢吡咯和200ppm吩噻嗪分别加入三口烧瓶中,搅拌均匀后,用平流泵连续泵入外径6mm、内径4mm和长400mm的管式反应器中,在温度110℃、压力1.0mpa和停留时间30min下进行甲醛与丙醛的mannich缩合反应。收集反应液进行气相色谱分析,并根据分析结果计算得到:丙醛转化率93.77%,甲基丙烯醛选择性96.90%和甲醛有效利用率90.86%。
74.实施例6
75.采用三聚甲醛解聚配制50%的甲醛溶液为原料。将1.000mol甲醛、1.000mol丙醛、0.010mol乙酸、0.006mol哌啶和250ppm三(1-羟基-2,2,6,6-四甲基哌啶-4-)亚磷酸酯(zj-705)分别加入三口烧瓶中,搅拌均匀后,用平流泵连续泵入外径6mm、内径4mm和长400mm的管式反应器中,在温度120℃、压力1.0mpa和停留时间30min下进行甲醛与丙醛的mannich缩合反应。收集反应液进行气相色谱分析,并根据分析结果计算得到:丙醛转化率92.57%,甲基丙烯醛选择性98.67%和甲醛有效利用率91.34%。
76.实施例7
77.采用37%的工业甲醛溶液(含约10%甲醇)为原料。将1.100mol甲醛、1.000mol丙醛、0.010mol丙酸、0.010mol二丙胺、100ppm对苯二酚和100ppm zj-701分别加入三口烧瓶中,搅拌均匀后,用平流泵连续泵入外径6mm、内径4mm和长400mm的管式反应器中,在温度125℃、压力1.2mpa和停留时间45min下进行甲醛与丙醛的mannich缩合反应。收集反应液进行气相色谱分析,并根据分析结果计算得到:丙醛转化率98.38%,甲基丙烯醛选择性98.01%和甲醛有效利用率87.66%。
78.实施例8
79.采用37%的工业甲醛溶液(含约10%甲醇)为原料。将1.100mol甲醛、1.000mol丙醛、0.006mol丁二酸和0.012mol二丙胺、100ppm对甲酚和100ppm zj-705分别加入三口烧瓶中,搅拌均匀后,用平流泵连续泵入外径6mm、内径4mm和长400mm的管式反应器中,在温度130℃、压力1.5mpa和停留时间45min下进行甲醛与丙醛的mannich缩合反应。收集反应液进行气相色谱分析,并根据分析结果计算得到:丙醛转化率98.44%,甲基丙烯醛选择性98.20%和甲醛有效利用率87.88%。
80.实施例9
81.采用37%的工业甲醛溶液(含约10%甲醇)为原料。将1.050mol甲醛、1.000mol丙醛、0.008mol丙二酸和0.010mol二丁胺、100ppm对苯二酚和100吩噻嗪分别加入三口烧瓶中,搅拌均匀后,用平流泵连续泵入外径6mm、内径4mm和长400mm的管式反应器中,在温度110℃、压力0.9mpa和停留时间60min下进行甲醛与丙醛的mannich缩合反应。收集反应液进行气相色谱分析,并根据分析结果计算得到:丙醛转化率98.22%,甲基丙烯醛选择性98.52%和甲醛有效利用92.16%。
82.实施例10
83.采用37%的工业甲醛溶液(含约10%甲醇)为原料。将1.050mol甲醛、1.000mol丙醛、0.005mol丁二酸和0.015mol二丁胺和250ppm对苯二酚分别加入三口烧瓶中,搅拌均匀后,用平流泵连续泵入外径6mm、内径4mm和长400mm的管式反应器中,在温度120℃、压力1.0mpa和停留时间60min下进行甲醛与丙醛的mannich缩合反应。收集反应液进行气相色谱分析,并根据分析结果计算得到:丙醛转化率98.78%,甲基丙烯醛选择性98.43%和甲醛有效利用率92.60%。
84.对比例5
85.试验过程、条件和各物质用量均同实施例10,区别在于,不添加阻聚剂对苯二酚。丙醛转化率99.12%,甲基丙烯醛选择性91.05%和甲醛有效利用率85.95%。
86.从实施例4~10和对比例5可知,采用连续管式反应工艺,所选择的有机胺-有机酸复合催化剂均表现出高的反应活性,而且添加阻聚剂有利于提高目标产物甲基丙烯醛选择性。
87.实施例11~12和对比例6~7为不同操作条件下反应产物的分离精制效果
88.实施例11
89.将实施例2的缩合反应产物收集,并送入油水分离罐中,在温度35℃下静置萃取时间5h,物料分为上下两层,下层水相物料直接排出;从油水分离罐中抽出上层油相物料,加入200ppm阻聚剂对苯二酚,混匀后送入脱轻组分塔,在压力50kpa、塔釜温度48℃、塔顶温度40℃和回流比40:1下精馏,塔顶采出丙醛物料;将脱轻组分塔塔釜物料送入产品精馏塔,在压力30kpa、塔釜温度60℃、塔顶温度35℃和回流比1:10下精馏,塔顶采出甲基丙烯醛产品,塔底为重组分。缩合反应产物通过分离精制,产品甲基丙烯醛纯度99.95%、分离收率96.06%。
90.实施例12
91.将实施例2的缩合反应产物收集,并送入油水分离罐中,在温度25℃下静置萃取时间2h,物料分为上下两层,下层水相物料直接排出;从油水分离罐中抽出上层油相物料,加
入300ppm阻聚剂对苯二酚,混匀后送入脱轻组分塔,在压力65kpa、塔釜温度52℃、塔顶温度45℃和回流比45:1下精馏,塔顶采出丙醛物料;将脱轻组分塔塔釜物料送入产品精馏塔,在压力50kpa、塔釜温度70℃、塔顶温度50℃和回流比1:20下精馏,塔顶采出甲基丙烯醛产品,塔底为重组分。缩合反应产物通过分离精制,产品甲基丙烯醛纯度99.93%、分离收率95.68%。
92.对比例6
93.将实施例2的缩合反应产物收集,并送入油水分离罐中,在温度45℃下静置萃取时间10h,物料分为上下两层,下层水相物料直接排出;从油水分离罐中抽出上层油相物料,加入200ppm阻聚剂对苯二酚,混匀后送入脱轻组分塔,在常压、塔釜温度103℃、塔顶温度65℃和回流比50:1下精馏,塔顶采出丙醛物料;将脱轻组分塔塔釜物料送入产品精馏塔,在压力常压、塔釜温度68℃、塔顶温度50℃和回流比1:8下精馏,塔顶采出甲基丙烯醛产品,塔底为重组分。缩合反应产物通过分离精制,产品甲基丙烯醛纯度98.90%、分离收率91.88%。
94.对比例7
95.将实施例2的缩合反应产物收集,并送入油水分离罐中,在温度15℃下静置萃取时间2h,物料分为上下两层,下层水相物料直接排出;从油水分离罐中抽出上层油相物料并直接送入脱轻组分塔,在20kpa、塔釜温度45℃、塔顶温度25℃和回流比30:1下精馏,塔顶采出丙醛物料;将脱轻组分塔塔釜物料送入产品精馏塔,在压力20kpa、塔釜温度60℃、塔顶温度35℃和回流比1:16下精馏,塔顶采出甲基丙烯醛产品,塔底为重组分。缩合反应产物通过分离精制,产品甲基丙烯醛纯度98.53%、分离收率88.56%。
96.从实施例11~12和对比例6~7可知,甲醛与丙醛的缩合反应产物,通过萃取分层实现油水分离、油相精馏脱出轻组分和重组分,可获得分离收率高于95%和纯度在99.9%以上的高品质甲基丙烯醛产品,但必须在产品分离精制过程中采取负压精馏并添加阻聚剂,以避免热敏性物料发生聚合而影响目标产品收率和纯度。
97.实施例13~18和对比例8为不同操作工况下反应及分离全流程放大试验
98.实施例13
99.按图1所示的流程,甲醛原料采用37%的工业甲醛溶液(含约10%甲醇),按摩尔配比甲醛:丙醛:丙酸:二丙胺=1.020:1.000:0.010:0.010分别将丙醛贮罐、催化剂罐和甲醛贮罐的物料加入至配料罐配制物料,然后加入250ppm对苯二酚并混匀,再连续送入外径10mm、内径8mm和长1000mm的管道式反应器中,在温度120℃、压力1.0mpa和停留时间45min下连续进行甲醛与丙醛的mannich缩合反应。将反应产物冷却至30℃并连续送入油水分离罐,静置萃取5h分层,下层水相物料直接排出体系;连续抽出上层油相物料并加入250ppm对苯二酚,再送入脱轻组分塔,在压力50kpa、塔釜温度48℃、塔顶温度40℃和回流比40:1下连续精馏,塔顶采出丙醛物料;脱轻组分塔塔釜物料送入产品精馏塔,在压力30kpa、塔釜温度60℃、塔顶温度35℃和回流比1:10下连续精馏,塔顶采出甲基丙烯醛产品,塔底重组分作为副产品回收。
100.对缩合反应液进行气相色谱分析,根据分析结果计算得到:缩合反应丙醛转化率96.18%,甲基丙烯醛选择性98.36%,甲醛有效利用率92.75%;通过对缩合反应产物的分离精制,以及对产品甲基丙烯醛含量的气相色谱分析和称重,计算得到:产品甲基丙烯醛纯度99.97%,分离收率96.78%。
101.对比例8
102.装置、物料配制及比例、缩合反应及反应产物的分离精制过程和操作条件均同实施例13,不同之处在于:在缩合反应和后续甲基丙烯醛的分离精制过程,均不加入阻聚剂对苯二酚。
103.缩合反应结果为:丙醛转化率99.36%,甲基丙烯醛选择性90.25%,甲醛有效利用率87.91%;反应产物分离精制结果为:产品甲基丙烯醛纯度99.52%、分离收率86.76%。
104.实施例14
105.装置、物料配制及比例、缩合反应及反应产物的分离精制过程和操作条件均同实施例13,不同之处在于:将脱轻组分塔塔顶采出的丙醛物料送回丙醛贮罐,用于配制反应物料。
106.缩合反应结果为:丙醛转化率98.10%,甲基丙烯醛选择性98.15%,甲醛有效利用率94.40%;反应产物分离精制结果为:产品甲基丙烯醛纯度99.97%、分离收率96.55%。
107.实施例15
108.装置、物料配制及比例、缩合反应及反应产物的分离精制过程和操作条件均同实施例13,不同之处在于:将所述脱轻组分塔塔顶采出的丙醛物料送回丙醛贮罐,并用于配制反应物料;同时将油水分离罐下层含催化剂、阻聚剂、甲醇和醛的水相物料,75%送回甲醛贮罐并加入三聚甲醛,通过解聚配制37%甲醛原料,其余25%作为废水排出体系。
109.缩合反应结果为:丙醛转化率99.28%,甲基丙烯醛选择性98.08%,甲醛有效利用率97.37%;反应产物分离精制结果为:产品甲基丙烯醛纯度99.95%、分离收率96.25%。
110.实施例16
111.按照图1所示的流程,采用三聚甲醛解聚配制50%的甲醛溶液,再按摩尔配比甲醛:丙醛:丁二酸:二乙胺=1.050:1.000:0.006:0.012加入至配料罐配制物料,加入250ppm对苯二酚并混匀,然后连续送入外径22mm、内径20mm和长2000mm的管道式反应器中,在温度110℃、压力0.8mpa和停留时间60min下连续进行甲醛与丙醛的mannich缩合反应。将反应产物冷却至35℃并连续送入油水分离罐,静置萃取6h分层,下层水相物料直接排出体系;连续抽出上层油相物料并加入250ppm对苯二酚,再送入脱轻组分塔,在压力70kpa、塔釜温度52℃、塔顶温度45℃和回流比45:1下连续精馏,塔顶采出丙醛物料;脱轻组分塔塔釜物料送入产品精馏塔,在压力50kpa、塔釜温度75℃、塔顶温度50℃和回流比1:15下连续精馏,塔顶采出甲基丙烯醛产品,塔底重组分作为副产品回收。
112.对缩合反应液进行气相色谱分析,根据分析结果计算得到:缩合反应丙醛转化率96.88%,甲基丙烯醛选择性98.56%,甲醛有效利用率90.94%;通过对缩合反应产物的分离精制,以及对产品甲基丙烯醛含量的气相色谱分析和称重,计算得到:产品甲基丙烯醛纯度99.96%,分离收率96.36%。
113.实施例17
114.装置、物料配制及比例、缩合反应及反应产物的分离精制过程和操作条件均同实施例16,不同之处在于:将所述脱轻组分塔塔顶采出的丙醛物料送回丙醛贮罐,并用于配制反应物料。
115.缩合反应结果为:丙醛转化率98.65%,甲基丙烯醛选择性98.36%,甲醛有效利用率92.41%;反应产物分离精制结果为:产品甲基丙烯醛纯度99.95%、分离收率96.28%。
116.实施例18
117.装置、物料配制及比例、缩合反应及反应产物的分离精制过程和操作条件均同实施例16,不同之处在于:将所述脱轻组分塔塔顶采出的丙醛物料送回丙醛贮罐,并用于配制反应物料;同时将所述油水分离罐下层含催化剂、阻聚剂、甲醇和醛的水相物料,60%送回甲醛贮罐并加入三聚甲醛,通过解聚配制50%浓度的甲醛原料,其余40%作为废水排出体系。
118.缩合反应结果为:丙醛转化率99.16%,甲基丙烯醛选择性98.12%,甲醛有效利用率97.30%;反应产物分离精制结果为:产品甲基丙烯醛纯度99.95%、分离收率96.10%。
119.上述所有实施例及对比例的原料摩尔配比、有机酸-有机碱催化剂组成及用量(酸或碱与原料的摩尔比)、阻聚剂种类及用量(阻聚剂占物料的质量比)、反应工艺条件汇总在表1,对应的原料甲醛来源及浓度、反应器形式及反应方式和装置规模、缩合反应结果见表2;实施例11~18及对比例6~8中产物分离精制的操作条件及甲基丙烯醛分离精制收率和产品纯度见表3。
120.表1各实施例及对比例的物料配比及反应工艺条件
121.[0122][0123]
表2各实施例和对比例的原料甲醛来源、反应方式及反应结果
[0124]
[0125][0126]
表3实施例11~18及对比例6~8的分离条件及甲基丙烯醛分离收率和纯度
[0127]
[0128][0129]
结合表1、表2和表3的数据,从实施例13~15和对比例8的全流程模试试验(与实施例1~12相比,放大10倍),以及实施例16~18的全流程中试试验(与实施例1~12相比,放大125倍)可知,甲醛与丙醛的mannich缩合反应及产物分离精制,几乎不存在工程放大效应,分别放大10倍和125倍的结果相当;在反应及分离过程中采用阻聚技术,可以减缓热敏性物料的聚合倾向,进行提高原料利用率;在将未反应完全的丙醛物料以及60~75%含催化剂、阻聚剂和少量醛的水相物料进行循环使用的情况下,丙醛转化率和甲醛有效利用率均得到提高,分别提高2~3%和4~7%,同时可以节省60~70%的催化剂和阻聚剂,以及减少60~70%的废水排放。因此,可以降低原料消耗、催化剂和阻聚剂消耗、能耗以及废水量,进而大幅降低生产成本。
[0130]
综上,所有实施例和对比例均表明:采用连续化的管道式反应工艺、高效分离精制(萃取分离-负压脱轻-产品精馏)工艺、物料循环工艺、热敏性物系阻聚技术以及新型高效的有机酸-有机胺组合催化剂,使甲醛和丙醛通过mannich缩合反应生产甲基丙烯醛技术得以实现,不仅克服了传统间歇釜式工艺效率低、催化剂用量大、废水量大、难以规模化生产等缺点,而且解决了微通道反应工艺存在的固有问题,如微通道反应器因微管过小,对于有
聚合倾向的热敏性物料体系,易堵塞而难以实现长周期运行,更难以实现物料循环,尤其是含催化剂、阻聚剂和微量聚合物或焦油的水相物料。此外,采用连续管道式反应工艺,操作条件较现有技术所采用微通道反应器的工艺条件更温和。
[0131]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。