460nm。
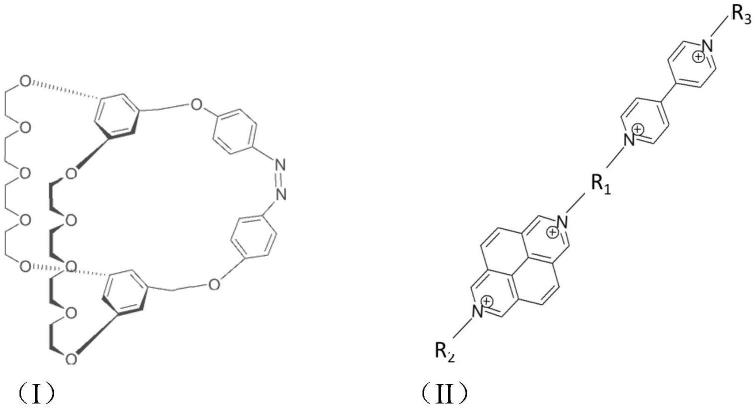
17.本发明还提供了所述基于双间苯-32-冠-10主体穴醚的分子机器的制备方法,包括以下步骤:将所述线性分子的六氟磷酸盐衍生物和双间苯-32-冠-10主体穴醚在乙腈溶液中以1:1摩尔比混合制备得到所述分子机器。
18.所述线性分子的六氟磷酸盐衍生物的合成方法包括以下步骤:
19.(i)将4,4’联吡啶与苄溴类化合物在有机溶剂中反应,反应得到的产物与nh4pf6作用得到中间体1-1;
20.(ii)将中间体1-1与2,7-氮杂芘在有机溶剂中反应,反应得到的产物与nh4pf6作用得到中间体1-2;
21.(iii)将中间体1-2与苄溴类化合物在有机溶剂中反应,反应得到的产物与nh4pf6作用得到所述线性分子的六氟磷酸盐衍生物;
22.所述的苄溴类化合物为对二苄溴或4,4'-双(溴甲基)-1,1'-联苯。
23.步骤(i)-(iii)中,有机溶剂为乙腈。
24.所述双间苯-32-冠-10主体穴醚的合成方法包括以下步骤:
25.(1)将3,5-二羟基苯甲酸甲酯和碳酸钾溶于有机溶剂中,再加入苄溴,反应得到中间体3-1;
26.(2)将中间体3-1、四甘醇双硫磺酸盐和碳酸钾溶于有机溶剂中,反应得到中间体3-2;
27.(3)将中间体3-2溶于混合溶剂中,加入催化剂反应得到中间体3-3;
28.(4)将中间体3-3、四甘醇双硫磺酸盐溶于有机溶剂中,再与溶于有机溶剂的碳酸钾混合,反应得到中间体3-4;
29.(5)将lialh4、中间体3-4在有机溶剂中反应得到中间体3-5;
30.(6)将中间体3-5溶于混合溶剂中,再加入pbr3反应,得到中间体3-6;
31.(7)将koh和对硝基苯酚在水中混合搅拌加热反应得到中间体3-7;
32.(8)将中间体3-6和中间体3-7溶于有机溶剂中,再与溶于有机溶剂的碳酸钾混合,反应得到所述双间苯-32-冠-10主体穴醚。
33.步骤(1)、(2)、(4)和(8)中,所述的有机溶剂为乙腈;
34.步骤(3)中,所述的混合溶剂为三氯甲烷和甲醇的混合溶剂;所述的催化剂为钯炭催化剂;
35.步骤(5)中,所述的有机溶剂为四氢呋喃;
36.步骤(6)中,所述的混合溶剂为乙醚和乙酸乙酯的混合溶剂。
37.与现有技术相比,本发明的有益效果在于:
38.(1)本发明提供的基于双间苯-32-冠-10主体穴醚的分子机器调控方便、具有基于光致结构变化的光响应性和输出信号,且制备方法简单、反应条件温和、原料易得。
39.(2)本发明采用双间苯-32-冠-10主体穴醚作为主体分子,双间苯-32-冠-10主体穴醚空间结构紧凑,使得主、客体组分间相互作用力更强,在形成和稳定索烃结构的过程中起到了重要的作用。
附图说明
40.图1为紫外吸收光谱图,其中,a为n,n
’‑
二甲基-2,7-氮杂芘盐2a,b为双间苯-32-冠-10主体穴醚trans-1溶液中加入等当量的客体n,n
’‑
二甲基-2,7-氮杂芘盐2a的溶液;c为b中溶液在350nm紫外光下照射1min;d为c中溶液在450nm下照射10min。
具体实施方式
41.下面结合附图与实施例,进一步阐明本发明。应理解,这些实施例仅用于说明本发明,而不用于限制本发明的范围。
42.实施例1线性分子的六氟磷酸盐衍生物合成
43.(1)化合物1.1的合成
[0044][0045]
将4,4’联吡啶(1.56g,10mmol)溶于50ml ch3cn中通过注射泵以2ml/h的速度加入到已回流的对二苄溴(10.0g,38.2mmol)的ch3cn溶液中。滴加结束后继续回流搅拌2小时。停止反应,待冷却后过滤出棕色沉淀,用500ml ch2cl2清洗滤液,干燥后融入2l水中,慢慢加入nh4pf6直到震荡后不再出现黄色絮状沉淀,将这些沉淀滤出,并用大量水清洗(100ml),真空干燥后得到了化合物1.1(浅灰色固体,3.4g,41.9%)。
[0046]
化合物1.1的鉴定
[0047]1h nmr(500mhz,cd3cn):9.48(d,4h,j=6.0hz),8.80(d,4h,j=6.0hz),7.62(d,4h,j=8.0hz),6.15(s,4h),4.67(s,4h)。
[0048]
(2)化合物1.2的合成
[0049][0050]
将化合物1.1(325mg,0.40mmol)与2,7-氮杂芘(60.0mg,0.30mmol)溶于50ml乙腈中,回流反应12小时。溶液颜色会逐渐变为黄色并有黄色沉淀出现。蒸去溶剂,将浓缩物溶于meoh和meno2的混合溶剂中搅拌,进行柱层析(硅胶,meoh/nh4cl/meno2,三者的体积比为20:2:1)。通过薄层层析确定并收集产物(产物有强荧光),浓缩含有产物的洗脱剂,然后溶
于少量热水中(5ml),一边震荡一边慢慢加入nh4pf6直到不再出现黄色絮状沉淀。将这些沉淀滤出,并用大量水清洗。干燥后得到化合物1.2(黄色,77mg,23.7%)。
[0051]
化合物1.2的鉴定
[0052]1h nmr(500mhz,cd3cn,室温)δ(ppm):4.62(s,2h),5.80(s,2h),5.84(s,2h),6.21(s,2h),7.47(s,4h),7.58(d,j=10hz,2h),7.69(d,j=10hz,2h),8.36(t,j=7.0hz,4h),8.51(d,j=9.0hz,2h),8.69(d,j=9.0hz,2h),8.95(t,j=7.0hz,4h),9.67(s,2h),9.85(s,2h)。
[0053]
(3)化合物1.3的合成
[0054][0055]
将化合物1.2(54.0mg,0.05mmol)和对二苄溴(1.00g,3.8mmol)溶于50ml ch3cn中,加热回流反应2小时。待冷却至室温,过滤得到黄色沉淀,用100ml乙醚清洗沉淀,然后将沉淀溶于少量热水(5ml),一边震荡一边慢慢加入nh4pf6直到不再出现黄色絮状沉淀,将这些沉淀滤出,并用大量水清洗。干燥后得到化合物1(黄色,56mg,79.4%)。
[0056]
化合物1的鉴定
[0057]1h nmr(500mhz,cd3cn,室温)δ(ppm):4.62(s,2h),5.81(s,2h),5.85(s,2h),6.25(s,2h),6.29(s,4h),7.48(s,4h),7.60(m,6h),7.40(d,j=8hz,2h),8.37(t,j=7.0hz,4h),8.83(t,j=2.0hz,4h),8.95(t,j=7.0hz,4h),9.94(s,4h)。
[0058]
实施例2双间苯-32-冠-10主体穴醚
[0059]
(1)化合物3.1的合成
[0060][0061]
将3,5-二羟基苯甲酸甲酯(150mmol,25.2g)和碳酸钾(100mmol,10.0g)溶于300ml无水乙腈(mecn)中,n2保护下加热至回流。再将苄溴(100mmol,17.1g)溶于100ml无水乙腈中,缓慢滴加(约4小时)到上述回流溶液中。让混合物在室温下搅拌10min。滴加结束后让反应液再继续搅拌4小时。蒸去乙腈并加入500ml ch2cl2溶剂反应混合物,将不溶物滤掉。蒸去ch2cl2进行柱层析(硅胶,石油醚/乙酸乙酯,(石油醚和乙酸乙酯的体积比为20:1)到(石油醚和乙酸乙酯的体积比为5:1))得到化合物3.1(白色固体,13.2g,分离产率51.2%)。
[0062]
化合物3.1的鉴定
[0063]1h nmr(400mhz,cd3cn):7.39-7.46(m,5h),7.28(s,1h),7.17(s,1h),6.70(s,1h),5.10(s,2h),3.92(s,3h)。
[0064]
(2)化合物3.2的合成
[0065][0066]
向化合物3.1(46.5mmol,12.0g),四甘醇双硫磺酸盐(23.2mmol,12.0g)和k2co3(100mmol,13.8g)中加入50ml乙腈(mecn)。n2保护下,加热回流反应24小时。冷却至室温,过滤并蒸去mecn,加入200ml ch2cl2溶解上步得到的混合物,再次过滤,蒸去进行柱层析(硅胶,石油醚/乙酸乙酯,(石油醚和乙酸乙酯的体积比为2:1)到(石油醚和乙酸乙酯的体积比为1:1))得到化合物3.2(黄色油状物,13.9g,分离产率88.8%)。
[0067]
化合物3.2的鉴定
[0068]1h nmr(400mhz,cd3cl3,22℃):δ3.68-3.73(m,8h),3.84(t,j=4.8hz,4h),5.06(s,4h),6.74(s,2h),7.21(s,2h),7.28(s,2h),7.34-7.40(m,10h)。
[0069]
(3)化合物3.3的合成
[0070][0071]
将化合物3.2(20.6mmol,13.9mg)溶于150ml chcl3和meoh的混合溶剂中(chcl3和meoh的体积比为1:1),加入500mg 10%pd/c催化剂,抽真空通入氢气加热室温搅拌反应。反应24h后,过滤并浓缩。得到化合物3.3(黄色油状物,9.2g,分离产率90.3%)。
[0072]
化合物3.3的鉴定
[0073]1h nmr(400mhz,cd3cl3,室温):δ3.66-3.68(m,8h),3.74(t,j=4.4hz,4h),3.84(s,6h),4.01(t,j=4.4hz,4h),6.10(t,j=4.0hz,2h),6.98(s,2h),7.10(s,2h),7.48(s,2h)。
[0074]
(4)化合物3.4的合成
[0075][0076]
n2保护下,在2l的三颈烧瓶中,加入k2co3(186mmol,25.7g)和1.5l mecn,加热至回流搅拌。将化合物3.3(18.6mmol,9.2g)和四甘醇双硫磺酸盐(18.6mmol,9.7g)溶于50ml mecn中,通过注射泵以1.5ml/h的速度加到上述的回流溶液中。滴加结束后,反应混合物在回流条件下继续搅拌3天。冷却至室温后,过滤并浓缩滤液。加入500ml ch2cl2溶解浓缩物并中再次过滤,蒸去ch2cl2,进行柱层析(硅胶,石油醚/乙酸乙酯,(石油醚和乙酸乙酯的体积
比为3:2)到(石油醚和乙酸乙酯的体积比为2:3)),得到化合物3.4(白色固体,6.9g,分离产率56.8%)。
[0077]
化合物3.4的鉴定
[0078]1h nmr(400mhz,cdcl3,室温):δ3.68-3.72(m,16h),3.85(t,j=4.8hz,4h),3.87(s,6h),4.10(t,j=4.8hz,4h),6.67(s,2h),7.15(d,j=4.8hz,4h)。
[0079]
(5)化合物3.5的合成
[0080][0081]
在250ml圆底烧瓶中加入lialh4(26.3mmol,1.00g),抽真空后氮气保护,在0℃下加入溶于150ml四氢呋喃(thf)的化合物3.4(8.43mmol,6.5g),反应24h后用乙酸乙酯淬灭,并用大量水稀释。用1m hcl中和后,用氯仿萃取。收集有机相饱和食盐水洗2次后用无水na2so4干燥。溶剂旋干后得到化合物3.5(白色固体,5.24g,分离产率88.1%)。
[0082]
化合物3.5的鉴定
[0083]1h nmr(400mhz,cdcl3,室温):δ2.43(s,2h),3.66-3.70(m,16h),3.81(t,j=4.4hz,8h),4.03(t,j=4.4hz,8h),4.54(d,j=2.8hz,4h),6.44(t,j=4.4hz,8h),6.48(d,j=2.0hz,4h)。
[0084]
(6)化合物3.6的合成
[0085][0086]
将化合物3.5(1.01mmol,0.60g)溶于70ml乙醚和10ml的乙酸乙酯的混合溶剂中。再将pbr3(6.06mmol,0.58ml)滴加到上述溶液中。室温下反应36小时。将反应体系中的白色沉淀过滤,用乙酸乙酯清洗两遍。最后在乙酸乙酯中重结晶得到化合物3.6(白色固体,0.65g,分离产率85%)。
[0087]
化合物3.6的鉴定:1h nmr(400mhz,cdcl3,22℃):δ3.67-3.73(m,16h),3.83(t,j=4.8hz,8h),4.06(t,j=4.8hz,8h),4.38(s,4h),6.42(t,j=2.4hz,2h),6.53(d,j=2.4hz,4h)。
[0088]
(7)化合物3.7的合成
[0089][0090]
将koh(760mmol,50g)、对硝基苯酚(72.0mmol,10g)和20ml水混合搅拌加热至20℃并保持1h。在慢慢将温度升至195-200℃,反应混合物会剧烈反应,有大量泡沫生成并呈深
棕色粘稠状。此时停止反应,将反应混合物溶入水中,再将这种深红色的溶液调制ph=3。用乙醚萃取,合并乙醚萃取物,加入无水硫酸钠干燥过夜,蒸去乙醚,将得到的产物在50%的乙醇水溶液中重结晶得到化合物3.7(黄色晶体,1.6g,分离产率17.2%)。
[0091]
化合物3.7的鉴定
[0092]1h nmr(400mhz,dmso-d6,22℃):δ10.10(s,2h),7.71(d,j=8.4hz,4h),7.71(d,j=8.8hz,4h),6.90(d,j=8.8hz,4h)。
[0093]
(8)化合物3.8(trans-1)的制备
[0094][0095]
氮气保护下,在1l的三颈烧瓶中,加入k2co3(20.0mmol,2.76g)和800ml mecn,加热至回流凉拌。将化合物3.6(1.04mmol,750mg)和化合物3.7(1.04mmol,222mg)溶于50ml mecn中,通过注射泵以1.5ml/h的速度加到上述已回流溶液中。滴加结束后,反应混合物在回流条件下继续搅拌3天。冷却至室温后,过滤并浓缩滤液。加入500ml ch2cl2溶解浓缩物并再次过滤,蒸去ch2cl2,进行柱层析(硅胶,石油醚/乙酸乙酯,(1:1,υ/υ)
→
(1:2,υ/υ)),得到化合物3.8(trans-1)(黄色固体,413mg,分离产率53.4%)。
[0096]
化合物3.8的鉴定
[0097]1h nmr(500mhz,cd3cocd3,室温):δ3.43-3.49(m,16h),3.66(t,j=4.5hz,8h),4.04(t,j=4.5hz,8h),5.31(s,4h),6.28(s,2h),6.51(d,j=2.0hz,4h),7.06(d,j=9.0hz,4h),7.64(d,j=9.0hz,4h)。
[0098]
13
c nmr(125mhz,acetone-d6,室温):δ 160.0,159.5,146.7,139.2,123.6,122.0,117.6,114.9,107.1,106.5,105.8,100.4,70.7,70.4,69.7,69.5,69.2,67.7,67.3。
[0099]
lresims:m/z 775.3[m h]
(64%),797.3[m na]
(100%),813.3[m k]
(35%).
[0100]
hrms:m/z calcd for[m k]
c42h50
n2o
12
h,813.2995,found 813.2961,error 4.0ppm。
[0101]
实施例3分子机器的合成
[0102]
将实施例1制得的线性分子的六氟磷酸盐衍生物和实施例2制得的双间苯-32-冠-10主体穴醚在乙腈溶液中以1:1摩尔比混合制备得到所述分子机器。
[0103]
通过核磁共振氢谱中氢的化学位移验证得到:在紫外光照下,双间苯-32-冠-10主体穴醚为顺式构型,由于更好的尺寸匹配效应,易于与式(iii)所示结构处络合,在可见光条件下,双间苯-32-冠-10主体穴醚为反式构型,易于与式(ⅳ)所示结构处络合;
[0104][0105]
样品分析
[0106]
如图1所示,在实施例1制得的反式结构的双间苯-32-冠-10主体穴醚trans-1的溶液中,加入等当量的客体n,n
’‑
二甲基-2,7-氮杂芘盐2a(结构式如下式所示,紫外吸收光谱图如图1中的a所示)后,在紫外光波长360nm附近的反式偶氮苯基团的特征吸收峰仍能对光发生响应:即紫外光照射后强度急剧下降,再用可见光照射时,强度又会升高(图1中的b-d)。由于客体2a本身吸收峰的影响,紫外光波长420nm左右与电子的n-π*跃迁相关吸收峰的变化不明显。与单独的主体trans-1相比,这种加入客体的体系,再使用可见光使其回复为反式时,效率明显下降。同样的条件下,用可见光照射10min,单独的trans-1其紫外光波长360nm处吸收峰强度回复了50%,而加入等当量客体后,仅回复了不到20%。这就表示客体的加入能够稳定的顺式结构。同时,核磁氢谱的结果证明顺式结构的双间苯-32-冠-10主体穴醚cis-1与客体2a存在良好的供体/受体相互作用。
[0107][0108]
客体n,n
’‑
二甲基-2,7-氮杂芘盐2a
[0109]
以上所述实施例对本发明的技术方案进行了详细说明,应理解的是以上所述仅为本发明的具体实施例,并不用于限制本发明,凡在本发明的原则范围内所做的任何修改、补充或类似方式替代等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
