1.本发明涉及有机合成技术领域,具体涉及一种5-羟基呋喃酮衍生物的制备方法。
背景技术:
2.呋喃酮结构广泛存在于具有生物活性的天然产物结构分子中。例如:从橐吾属植物中分离得到的艾里莫芬内酯(eremophilanolides)含有呋喃酮结构(chem.biodiversity 2016,13,645;nat.prod.rep.2006,23,699),该类化合物也被作为中草药应用于止咳和活血化瘀(mini-rev.med.chem.2014,14,664)。因此,开发简便高效的制备含有5-羟基呋喃酮衍生物结构的方法具有重要的工业生产和科学研究价值。
3.现有的制备5-羟基呋喃酮衍生物的方法以环状脂肪酮化合物与2-氯-2-甲氧基乙酸甲酯或1,1-二甲氧基丙酮为主,其中2-氯-2-甲氧基乙酸甲酯或1,1-二甲氧基丙酮需要通过多步反应制备得到(synthesis 1979,434;tetrahedron lett.2015,56,5545;j.org.chem.2004,69,9100),且存在产率较低、原料难得,副反应多等缺点。目前,尚无一种高效简便的制备5-羟基呋喃酮衍生物的方法。
技术实现要素:
4.针对以上问题,本发明提供了一种5-羟基呋喃酮衍生物的制备方法,该方法具有反应条件温和、操作简单、原料易得、官能团兼容性好、底物适用范围广的优点。
5.为了实现上述目的,本发明采用的技术方案如下:
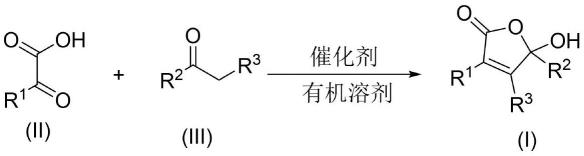
6.一种5-羟基呋喃酮衍生物的制备方法,具体包括以下步骤:在有机溶剂中依次加入式(ii)所示的α-酮酸化合物、式(iii)所示的脂肪酮化合物和催化剂,在一定反应温度下进行反应,通过柱层析色谱纯化即可制备得到5-羟基呋喃酮衍生物(i),其反应结构式如下所示;
[0007][0008]
其中,r1独立地选自苯基、取代苯基、萘基、噻吩基、c1-c6烷基、c1-c6烯基中的任意一种;r2和r3独立地选自氢原子、c1-c6烷基、c1-c6烯基中的任意一种。
[0009]
其中,c1-c6烷基是指具有1-6个碳原子的直链或支链烷基,其包括:甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、异戊基、正己基环己基等。
[0010]
c1-c6烯基是指具有1-6个碳原子的直链或支链含有碳碳双键的取代基,其包括:乙烯基、丙烯基、丁烯基、戊烯基、己烯基、环己烯基。
[0011]
优选地,有机溶剂包括:甲苯、氟苯、三氟甲苯、氯苯、苯、二甲苯、四氢呋喃、甲醇、乙醇、乙腈、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜和n-甲基吡咯烷酮中的任意一种。
[0012]
优选地,催化剂包括:三氟化硼乙醚、三氟甲磺酸酐、对甲苯磺酸、氯化铁、三氟甲磺酸铋、三氟甲磺酸锌、三氟甲磺酸钇、三氟甲磺酸铜、氯化铝、四氯化钛中的任意一种。
[0013]
优选地,α-酮酸化合物(ii)与脂肪酮化合物(iii)的摩尔比为1:(1-3);所述α-酮酸化合物(ii)与催化剂的摩尔比为1:(0.05-1);所述α-酮酸化合物(ii)与有机溶剂的用量比为1mmol:(2-15)ml。
[0014]
优选地,反应温度为25-120℃,时间为0.5-12小时。
[0015]
优选地,柱层析色谱提纯所用的洗脱液为石油醚与乙酸乙酯的混合溶剂,所述石油醚:乙酸乙酯的体积比为(1~10):1。
[0016]
本发明有益效果:
[0017]
1、本发明采用廉价易得的α-酮酸化合物、脂肪酮化合物为原料,以三氟化硼乙醚为催化剂制备得到5-羟基呋喃酮衍生物。
[0018]
2、本发明可在空气条件下操作,对氧气不敏感,反应条件温和、操作简单。
[0019]
3、本发明具有官能团兼容性好、后处理简单、原子经济性高等优点。
[0020]
4、本发明为5-羟基呋喃酮衍生物的制备提供了新的合成路线,制备所得到的5-羟基呋喃酮衍生物可在活性药物中间体领域中发挥重要作用,在工业生产和科学研究上具有较大的应用价值和潜力。
具体实施方式
[0021]
以下通过具体实施方式对本发明的技术方案进行进一步的说明和描述,但本发明的实施方式不限于此。
[0022]
实施例1:
[0023][0024]
向甲苯中加入上式苯甲酰甲酸(ii)、环己酮化合物(iii)、三氟硼酸乙醚(bf3·
et2o),然后在40℃温度下搅拌密封反应2小时。
[0025]
其中,苯甲酰甲酸(ii)和环己酮化合物(iii)的摩尔比为1:2;苯甲酰甲酸(ii)与三氟化硼乙醚(bf3·
et2o)的摩尔比为1:0.2;苯甲酰甲酸(ii)与甲苯的比例为1mmol:4ml。
[0026]
反应结束后,向反应体系自中加入等体积比的乙酸乙酯和饱和食盐水的混合液,振荡萃取3次,收集有机层、干燥,旋转蒸发浓缩,得粗产物,将粗产物经300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1:20,即得到外观为白色固体的目标产物式(i)化合物(c
14h14
o3)。
[0027]
对本实施例得到的式(i)化合物(c
14h14
o3)进行核磁共振分析,结果为:1h nmr(400mhz,氘代氯仿cdcl3)δ7.44-7.34(m,5h),4.31(s,1h),2.97-2.94(m,1h),2.53-2.45(m,2h),2.03-1.91(m,1h),1.86-1.74(m,2h),1.64-1.55(m,1h),1.37-1.24(m,1h).
[0028]
13
c nmr(100mhz,氘代氯仿cdcl3)δ170.9,162.2,129.2,128.9(2c),128.6,128.5(2c),124.4,103.2,38.3,26.9,25.6,22.0.
[0029]
hrms m/z(esi)calcd for c
14h14
o3,(m h)
231.1016;found 231.1014.
[0030]
经测算:式(i)化合物(c
14h14
o3)的产率为86%,熔点:102
–
104℃。
[0031]
实施例2:
[0032][0033]
向甲苯中加入上式苯甲酰甲酸(ii)、四氢吡喃酮化合物(iii)、三氟硼酸乙醚(bf3·
et2o),然后在40℃温度下搅拌密封反应2小时。
[0034]
其中,苯甲酰甲酸(ii)和四氢吡喃酮化合物(iii)的摩尔比为1:2;苯甲酰甲酸(ii)与三氟化硼乙醚(bf3·
et2o)的摩尔比为1:0.2;苯甲酰甲酸(ii)与甲苯的比例为1mmol:4ml。
[0035]
反应结束后,向反应体系自中加入等体积比的乙酸乙酯和饱和食盐水的混合液,振荡萃取3次,收集有机层、干燥,旋转蒸发浓缩,得粗产物,将粗产物经300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1:20,即得到外观为无色油状液体的目标产物式(i)化合物(c
13h12
o4)。
[0036]
对本实施例得到的式(i)化合物(c
13h12
o4)进行核磁共振分析,结果为:1h nmr(400mhz,氘代氯仿cdcl3)δ7.45-7.31(m,5h),4.75(d,j=13.2hz,1h),4.41-4.38(m,2h),4.00-3.96(m,1h),3.86-3.80(m,1h),2.37(d,j=13.4hz,1h),2.10-1.99(m,1h).
[0037]
13
c nmr(100mhz,氘代氯仿cdcl3)δ170.3,154.8,129.5,128.9(2c),128.7(2c),128.1,126.1,100.9,64.6,62.7,40.0.
[0038]
hrms m/z(esi)calcd for c
13h12
o4,(m h)
233.0808;found 233.0806.
[0039]
经测算:式(i)化合物(c
13h12
o4)的产率为60%。
[0040]
实施例3:
[0041][0042]
向甲苯中加入上式苯甲酰甲酸(ii)、环庚酮化合物(iii)、三氟硼酸乙醚(bf3·
et2o),然后在40℃温度下搅拌密封反应2小时。
[0043]
其中,苯甲酰甲酸(ii)和环庚酮化合物(iii)的摩尔比为1:2;苯甲酰甲酸(ii)与三氟化硼乙醚(bf3·
et2o)的摩尔比为1:0.2;苯甲酰甲酸(ii)与甲苯的比例为1mmol:4ml。
[0044]
反应结束后,向反应体系自中加入等体积比的乙酸乙酯和饱和食盐水的混合液,振荡萃取3次,收集有机层、干燥,旋转蒸发浓缩,得粗产物,将粗产物经300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1:20,即得到外观为白色固体的目标产物式(i)化合物(c
15h16
o3)。
[0045]
对本实施例得到的式(i)化合物(c
15h16
o3)进行核磁共振分析,结果为:1h nmr
(400mhz,氘代氯仿cdcl3)δ7.44-7.32(m,5h),4.55(s,1h),2.84-2.63(m,2h),2.34(ddd,j=14.2,6.1,2.7hz,1h),1.99-1.55(m,6h),1.46-1.34(m,1h).
[0046]
13
c nmr(100mhz,氘代氯仿cdcl3)δ171.3,164.2,129.6,128.9(2c),128.5,128.3(2c),127.1,107.7,38.0,28.5,26.6,25.6,23.5.
[0047]
hrms m/z(esi)calcd for c
15h16
o3,(m h)
245.1172;found 245.1171.
[0048]
经测算:式(i)化合物(c
15h16
o3)的产率为79%,熔点:84
–
86℃。
[0049]
实施例4:
[0050][0051]
向甲苯中加入上式4-甲基苯甲酰甲酸(ii)、丙酮化合物(iii)、三氟硼酸乙醚(bf3·
et2o),然后在40℃温度下搅拌密封反应2小时。
[0052]
其中,4-甲基苯甲酰甲酸(ii)和丙酮化合物(iii)的摩尔比为1:2;4-甲基苯甲酰甲酸(ii)与三氟化硼乙醚(bf3·
et2o)的摩尔比为1:0.2;4-甲基苯甲酰甲酸(ii)与甲苯的比例为1mmol:4ml。
[0053]
反应结束后,向反应体系自中加入等体积比的乙酸乙酯和饱和食盐水的混合液,振荡萃取3次,收集有机层、干燥,旋转蒸发浓缩,得粗产物,将粗产物经300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1:20,即得到外观为白色固体的目标产物式(i)化合物(c
11h10
o3)。
[0054]
对本实施例得到的式(i)化合物(c
11h10
o3)进行核磁共振分析,结果为:1h nmr(400mhz,氘代氯仿cdcl3)δ7.78-7.75(m,2h),7.40-7.33(m,3h),7.30(s,1h),4.41(s,1h),1.73(s,3h).
[0055]
13
c nmr(100mhz,氘代氯仿cdcl3)δ170.0,146.4,132.5,129.8,128.6(3c),127.4(2c),103.7,24.7.
[0056]
hrms m/z(esi)calcd for c
11h10
o3,(m h)
191.0703;found 191.0705.
[0057]
经测算:式(i)化合物(c
11h10
o3)的产率为59%,熔点:91-93℃。
[0058]
实施例5:
[0059][0060]
向甲苯中加入上式2-(萘-2-基)-2-氧代乙酸(ii)、环己酮化合物(iii)、三氟硼酸乙醚(bf3·
et2o),然后在40℃温度下搅拌密封反应2小时。
[0061]
其中,2-(萘-2-基)-2-氧代乙酸(ii)和卤代炔烃化合物(iii)的摩尔比为1:2;2-(萘-2-基)-2-氧代乙酸(ii)与三氟化硼乙醚(bf3·
et2o)的摩尔比为1:0.2;2-(萘-2-基)-2-氧代乙酸(ii)与甲苯的比例为1mmol:4ml。
[0062]
反应结束后,向反应体系自中加入等体积比的乙酸乙酯和饱和食盐水的混合液,振荡萃取3次,收集有机层、干燥,旋转蒸发浓缩,得粗产物,将粗产物经300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1:20,即得到外观为白色固体的目标产物式(i)化合物(c
18h16
o3)。
[0063]
对本实施例得到的式(i)化合物(c
18h16
o3)进行核磁共振分析,结果为:1h nmr(400mhz,氘代氯仿cdcl3)δ7.89(s,1h),7.78-7.73(m,3h),7.51-7.38(m,3h),4.70(s,1h),2.98(d,j=13.7hz,1h),2.60-2.44(m,2h),2.00-1.89(m,1h),1.82-1.75(m,2h),1.63-1.55(m,1h),1.37-1.17(m,1h).
[0064]
13
c nmr(100mhz,氘代氯仿cdcl3)δ171.3,162.7,133.0,132.9,128.6,128.2,128.0,127.6,126.6,126.6,126.2,126.1,124.3,103.5,38.2,26.9,25.8,22.0.
[0065]
hrms m/z(esi)calcd for c
18h16
o3,(m h)
281.1172;found 281.1170.
[0066]
经测算:式(i)化合物(c
18h16
o3)的产率为85%,熔点:135-137℃。
[0067]
实施例6:
[0068][0069]
向甲苯中加入上式4-甲基苯基乙醛酸化合物(ii)、环己酮化合物(iii)、三氟硼酸乙醚(bf3·
et2o),然后在40℃温度下搅拌密封反应2小时。
[0070]
其中,4-甲基苯基乙醛酸化合物(ii)和环己酮化合物(iii)的摩尔比为1:2;4-甲基苯基乙醛酸化合物(ii)与三氟化硼乙醚(bf3·
et2o)的摩尔比为1:0.2;4-甲基苯基乙醛酸化合物(ii)与甲苯的比例为1mmol:4ml。
[0071]
反应结束后,向反应体系自中加入等体积比的乙酸乙酯和饱和食盐水的混合液,振荡萃取3次,收集有机层、干燥,旋转蒸发浓缩,得粗产物,将粗产物经300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1:20,即得到外观为白色固体的目标产物式(i)化合物(c
15h16
o3)。
[0072]
对本实施例得到的式(i)化合物(c
15h16
o3)进行核磁共振分析,结果为:1h nmr(400mhz,氘代氯仿cdcl3)δ7.32(d,j=8.0hz,2h),7.19(d,j=7.8hz,2h),4.23(s,1h),2.95(d,j=13.7hz,1h),2.52-2.42(m,2h),2.00-1.94(m,1h),1.82-1.75(m,2h),1.62-1.55(m,1h),1.35-1.24(m,1h).
[0073]
13
c nmr(100mhz,氘代氯仿cdcl3)δ171.0,161.5,138.6,129.1(2c),128.8(2c),126.3,124.3,103.2,38.2,26.9,25.6,22.1,21.3.
[0074]
hrms m/z(esi)calcd for c
15h16
o3,(m h)
245.1172;found 245.1175.
[0075]
经测算:式(i)化合物(c
15h16
o3)的产率为84%,熔点:131-133℃。
[0076]
实施例7:
[0077][0078]
向甲苯中加入上式4-溴苯基乙醛酸化合物(ii)、环己酮化合物(iii)、三氟硼酸乙醚(bf3·
et2o),然后在40℃温度下搅拌密封反应2小时。
[0079]
其中,4-溴苯基乙醛酸化合物(ii)和环己酮化合物(iii)的摩尔比为1:2;4-溴苯基乙醛酸化合物(ii)与三氟化硼乙醚(bf3·
et2o)的摩尔比为1:0.2;4-溴苯基乙醛酸化合物(ii)与甲苯的比例为1mmol:4ml。
[0080]
反应结束后,向反应体系自中加入等体积比的乙酸乙酯和饱和食盐水的混合液,振荡萃取3次,收集有机层、干燥,旋转蒸发浓缩,得粗产物,将粗产物经300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1:20,即得到外观为白色固体的目标产物式(i)化合物(c
14h13
bro3)。
[0081]
对本实施例得到的式(i)化合物(c
14h13
bro3)进行核磁共振分析,结果为:1h nmr(400mhz,氘代氯仿cdcl3)δ7.55-7.49(m,2h),7.32-7.25(m,2h),3.95(s,1h),2.96-2.87(m,1h),2.56-2.43(m,2h),2.08-1.96(m,1h),2.04-1.99(m,2h),1.68-1.57(m,1h),1.42-1.25(m,1h).
[0082]
13
c nmr(100mhz,氘代氯仿cdcl3)δ170.5,162.6,131.7(2c),130.5(2c),128.0,123.5,123.1,103.3,38.3,26.9,25.7,22.0.
[0083]
hrms m/z(esi)calcd for c
14h13
bro3,(m h)
309.0121;found 309.0119.
[0084]
经测算:式(i)化合物(c
14h13
bro3)的产率为80%,熔点:168-170℃。
[0085]
实施例8:
[0086][0087]
向甲苯中加入上式2-噻吩乙醛酸化合物(ii)、环己酮化合物(iii)、三氟硼酸乙醚(bf3·
et2o),然后在40℃温度下搅拌密封反应2小时。
[0088]
其中,2-噻吩乙醛酸化合物(ii)和环己酮化合物(iii)的摩尔比为1:2;2-噻吩乙醛酸化合物(ii)与三氟化硼乙醚(bf3·
et2o)的摩尔比为1:0.2;2-噻吩乙醛酸化合物(ii)与甲苯的比例为1mmol:4ml。
[0089]
反应结束后,向反应体系自中加入等体积比的乙酸乙酯和饱和食盐水的混合液,振荡萃取3次,收集有机层、干燥,旋转蒸发浓缩,得粗产物,将粗产物经300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1:20,即得到外观为白色固体的目标产物式(i)化合物(c
12h12
so3)。
[0090]
对本实施例得到的式(i)化合物(c
12h12
so3)进行核磁共振分析,结果为:1h nmr(400mhz,氘代氯仿cdcl3)δ7.64(d,j=3.7hz,1h),7.39(d,j=5.1hz,1h),7.08(dd,j=
4.9,3.8hz,1h),3.92(s,1h),3.28-3.23(m,1h),2.61-2.42(m,2h),2.07-2.02(m,1h),1.89-1.72(m,2h),1.67-1.52(m,1h),1.46-1.29(m,1h).
[0091]
13
c nmr(100mhz,氘代氯仿cdcl3)δ169.8,158.5,130.7,128.5,127.3,126.9,118.2,103.3,38.3,26.7,26.2,22.0.
[0092]
hrms m/z(esi)calcd for c
12h12
so3,(m h)
237.0580;found 237.0582.
[0093]
经测算:式(i)化合物(c
12h12
so3)的产率为72%,熔点:70-72℃。
[0094]
综上所述,本发明是以α-酮酸化合物、脂肪酮化合物作为原料,以廉价易得的酸为催化剂制备得到5-羟基呋喃酮衍生物,该方法具有反应产率高、操作简单、原子经济性高、官能团兼容性好,底物适用范围广的优点。本发明为5-羟基呋喃酮衍生物的制备提供了全新的路线。
[0095]
需要说明的是,本发明未详述之处,均为本领域技术人员的公知技术。
[0096]
上述实施例仅用来进一步说明本发明的一种5-羟基呋喃酮衍生物的制备方法,但本发明并不局限于实施例,凡是根据本发明的技术实质,对以上实施例所作的等同变化与修饰皆包括在本发明技术方案的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
