1.本发明涉及有机合成领域,具体涉及一种在常温条件下以紫光作为光源,使用偶氮芳基保护的四氢异喹啉化合物(i)与苄或烯丙基溴化物(ii)作为底物,实现了可见光诱导偶氮芳基保护的四氢异喹啉化合物的α-烯丙或苄基化反应,去合成1-苄或烯丙基3,4-二氢异喹啉类产物(iii)。
背景技术:
2.异喹啉是包含有苯并吡咯的双环结构,氮原子位于吡啶环的2位,根据不饱和度可以分为异喹啉、二氢异喹啉、四氢异喹啉三种形式。在种类繁多的天然氮杂环生物碱家族中,四氢异喹啉类化合物,尤其是c1官能团化的四氢异喹啉生物碱分布十分广泛在罂粟科、小檗科、防已科、毛莨科、海洋动植物等众多生物中都能发现这类化合物的身影(e.m.shamma,2012;vol.25.)。这些四氢异喹啉生物碱往往具有显著而且多样的生物活性,例如从粉防已中提取的双苄基异喹啉生物碱粉防已碱,几十年来一直被用于治疗肿瘤、高血压、真菌感染等疾病(a)w.a.creasey,biochem.pharmacol.1976,25,1887-1891;b)y-j,chen,acta.pharmacol.sin.2002,23,1102-1106;c)l.x.zhao,l.de-dong,h.dan-dan,h.gan-hai,y.lan,w.yan,j.yuan-ying,c.tom,plos one,8,e79671;d)b.c.liu,y.x.he,q.miao,h.h.wang,b.r.you,biomed.environ.sci.1994,7,199-204.)。莲花叶片中分离得到的(r)-乌药碱能够抑制人t淋巴细胞h9中hiv病毒复制(y.kashiwada,a.aoshima,y.ikeshiro,y.p.chem,h.furukawa,m.itoigawa,t.fujioka,k.mihashi,l.m.cosentino,s.l.morris-natschke,k.h.lee,biorg.med.chem.2005,13,443-448)。延胡索乙素作为多巴胺受体拮抗剂(h.chu,g.jin,e.friedman,x.zhen,cell.mol.neurobiol.2008,28,491-499)。
3.四氢异喹啉是众多生理活性天然产物的核心骨架,对四氢异喹啉骨架进行修饰和改造已经成为新药研发的热门领域。一些人工合成的异喹啉类化合物也表现出了良好的生物活性。c1-吲哚取代的四氢异喹啉化合物索利那新是竞争性毒覃碱抑制剂,临床上用于缓解膀胱过度活动症(a.ohtake,m.ukai,t.hatanaka,s.kobayashi,k.ikeda,s.sato,k.miyata,m.sasamata,eur.j.pharmacol.2004,492,243-250.)。α-吲哚异喹啉类似物ibr2是rad51的小分子抑制剂(a)j.zhu,l.zhou,g.wu,h.konig,x.lin,g.li,x.-l.qiu,c.-f.chen,c.-m.hu,e.goldblatt,r.bhatia,a.r.chamberlin,p.-l.chen,w.-h.lee,embo mol.med.2013,5,353-365;b)t.-w.chung,y.-t.hung,t.thikekar,v.v.paike,f.y.lo,p.-h.tsai,m.-c.liang,c.-m.sun,acs comb.sci.2015,17,442-451;c)j.zhu,h.chen,x.e.guo.x.-l.qiu,c.-m.hu,a.r.chamberlin,w.-h.lee,eur,j.med.chem.2015,96,196-208.)。
4.叔胺的α-烯丙基烷基化反应是制备高烯丙基胺特别有用和具有挑战性的反应,而高烯丙基胺常被用作合成药物和天然产物的中间体(a)s.a.lawrence,amines:synthesis,properties and applications.cambridge university press,cambridge,2004;b)
t.c.nugent,chiral amine synthesis:methods,developments and applications,wiley-vch,weinheim,2010;c)c.o.puentes,v.kouznetsov,j.heterocycl.chem.2002,39,595-614.)。传统的叔胺直接α-烯丙基化方法主要是基于交叉脱氢偶联(cdc)策略(a)z.li and c.-j.li,j.am.chem.soc.2004,126,11810-11811;b)c.-j.li,acc.chem.res.2009,42,335-344.for applications of cdc inα-allylation of tertiary amines,see:c)g.kumaraswamy,a.murthyand a.pitchaiah,j.org.chem.2010,75,3916-3919;d)e.boess and m.klussmann,j.am.chem.soc.2011,133,8106-8109;e)j.w.tucker,y.zhang,t.f.jamison and c.r.j.stephenson,angew.chem.int.ed.2012,51,4144-4147;f)w.-j.yoo,a.tanoue and s.kobayashi,asian j.org.chem.2014,3,1066-1069;g)j.p.barham,m.p.john and j.a.murphy,beilstein j.org.chem.2014,10,2981-2988;h)l.and s.blechert,adv.synth.catal.2014,356,2825-2829;(i)t.-t.wang,m.schrempp,a.o.schiemann and d.menche,org.lett.2015,17,3982-3985;j)j.wang and s.yang,tetrahedron lett.2016,57,3444-3448;k)t.t.chen and c.cai,synlett.2017,28,1368-1372.)。相比之下,我们利用可见光诱导芳基偶氮基保护的四氢异喹啉与烯丙基/苄溴的烯丙基和苄基化反应,不需要水敏亲核试剂或化学计量学氧化剂,在温和条件下合成了1-苄/烯丙基3,4-二氢异喹啉类产物。
技术实现要素:
5.本发明克服了芳基三氮烯csp
3-h键活化的诸多缺点(如需当量的氧化剂氧化),本发明在仅需当量的碱,无促进剂,氮气氛围下,绿色高效地实现了交叉偶联反应去合成1-苄或烯丙基3,4-二氢异喹啉类产物。
6.本发明使用环境友好、可见光催化底物分子从而得到eda复合物,然后再自由基和自由基偶联。以制备简单、来源广泛的偶氮芳基保护的四氢异喹啉作为原料,实现可见光诱导偶氮芳基保护的四氢异喹啉化合物的α-烯丙或苄基化反应。
7.通过这种方法制备得到了如下反应式中1-(2-甲烯丙基)-3,4-二氢异喹啉(iii);
8.其中,所述反应过程如下反应式所示;
[0009][0010]
其中
[0011]
苄或烯丙基溴化物(ii)中虚线表示此处为无苯环或有苯环二种情形的溴化物,无苯环时溴化物为烯丙基溴化物,有苯环时溴化物为苄基溴化物;而1-苄或烯丙基3,4-二氢异喹啉类化合物(iii)中虚线表示此处为无苯环或有苯环二种情形的化合物,无苯环时其为1-烯丙基3,4-二氢异喹啉类化合物,有苯环时其为1-苄基3,4-二氢异喹啉类化合物;
[0012]
r、r1分别独立地表示在苯环邻位、间位、对位等中的一种或二种以上不同位置的取代基或为氢的无取代基,取代基的个数分别为1-5个,优选1-2个,其具体可分别独立地为
甲基、异丙基、氯、溴、碘、甲氧基中的一种或二种以上;r2表示为氢、甲基中的一种或二种;
[0013]
优选:r为氢、溴、甲氧基中的一种或二种以上,取代基的个数为1-5个,优选1-2个;r1为氢、2-甲基、3-甲基、4-甲基、4-异丙基、4-氯、4-溴中的一种或二种以上,取代基的个数为1-5个,优选1-2个;r2为氢、甲基中的一种或二种。
[0014]
如以上反应式,本发明一种在常温条件下以紫光作为光源,使用偶氮芳基保护的四氢异喹啉化合物(i)与苄或烯丙基溴化物(ii)作为底物,实现了可见光诱导偶氮芳基保护的四氢异喹啉化合物的α-烯丙或苄基化反应,去合成1-苄或烯丙基3,4-二氢异喹啉类产物(iii)。
[0015]
本发明中,所述反应中,反应光源为390-395nm紫光。
[0016]
本发明中,所述反应中,反应碱为k2co3。
[0017]
本发明中,所述反应中,反应添加剂为18-crown-6。
[0018]
本发明中,所述反应中,反应温度为25℃。
[0019]
本发明中,所述反应溶剂为ch3cn。
[0020]
本发明中,所述反应反应时间为36-48h。优选地,反应时间为36h。
[0021]
本发明合成反应包括以下步骤:
[0022]
偶氮芳基保护的四氢异喹啉的合成:
[0023][0024]
根据先前制备文献报道(l.g.margaret,h.b.david,m.w.willard.j.org.chem.1993,58,2104-2109.),在100ml的三口烧瓶中,加入芳基苯胺(10mmol),然后在0℃下将2ml的质量浓度12.0mol/l浓盐酸入三口烧瓶中。在50ml的烧杯中加入亚硝酸钠0.76g(11mmol),再加入5ml的蒸馏水使其溶解,然后用恒压滴液漏斗将亚硝酸钠水溶液逐滴加入到对甲基苯胺的盐酸溶液中,在0℃下继续搅拌10min,得芳基重氮盐溶液;在50ml烧杯中加入四氢异喹啉(10mmol)和1.2mol/l的碳酸钾溶液(1.6585g溶于10ml水),搅拌均匀后一次性将上述芳基重氮盐溶液加入其中,在0℃搅拌反应半小时后出现大量黄色固体,用布氏漏斗抽滤并用冰水洗涤固体,得粗产品,将无水乙醇加热到60℃,将粗产品转移至圆底烧瓶中,滴加60℃无水乙醇重结晶,得到淡黄色固体产物,真空干燥称重并计算产率。
[0025]
1-(2-甲烯丙基)-3,4-二氢异喹啉(iii)的合成:
[0026]
在石英管中依次加入(e)-2-(对甲苯基二氮烯)-1,2,3,4-四氢异喹啉(i)(x mmol)、2-甲基-3-溴丙烯(y mmol)、k2co3(z mmol)、18-crown-6(m mmol)以及溶剂dma(n ml),在n2氛围以及紫光(390-395nm)照射条件下搅拌36h,得到上述反应产物1-(2-甲烯丙基)-3,4-二氢异喹啉。
[0027]
本发明克服了偶氮芳基保护的四氢异喹啉csp
3-h键活化的诸多缺点(如需当量的氧化剂氧化),本发明在仅需光诱导催化,无促进剂,氮气氛围下,绿色高效地实现了偶氮芳
基保护的四氢异喹啉与苄/烯丙基溴化物去合成1-苄或烯丙基3,4-二氢异喹啉类化合物。本发明使用的各原料均为工业化商品,原料便宜易得、来源广泛、简单易得,来源广泛;制备简单、价格便宜、结构稳定、污染小、绿色环保;反应绿色、操作简单。
附图说明
[0028]
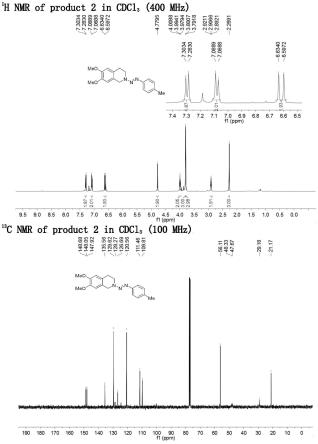
图1是实施例2产物(e)-6,7-二甲氧基-2-(对甲苯基二氮烯基)-1,2,3,4-四氢异喹啉的核磁氢碳谱图;
[0029]
图2是实施例3产物(e)-6溴-2-(对甲苯基二氮烯基)-1,2,3,4-四氢异喹啉的核磁氢碳谱图;
[0030]
图3是实施例4产物1-(2-甲烯丙基)-3,4-二氢异喹啉的核磁氢碳谱图;
[0031]
图4是实施例5产物1-苄基-3,4-二氢异喹啉的核磁氢碳谱图;
[0032]
图5是实施例6产物1-(4-甲基苄基)-3,4-二氢异喹啉的核磁氢碳谱图;
[0033]
图6是实施例7产物1-(3-甲基苄基)-3,4-二氢异喹啉的核磁氢碳谱图;
[0034]
图7是实施例8产物1-(2-甲基苄基)-3,4-二氢异喹啉的核磁氢碳谱图;
[0035]
图8是实施例9产物1-(4-异丙基苄基)-3,4-二氢异喹啉的核磁氢碳谱图;
[0036]
图9是实施例10产物1-(4-氯苄基)-3,4-二氢异喹啉的核磁氢碳谱图;
[0037]
图10是实施例11产物1-(4-溴苄基)-3,4-二氢异喹啉的核磁氢碳谱图;
[0038]
图11是实施例12产物1-(1-苯乙基)-3,4-二氢异喹啉的核磁氢碳谱图;
[0039]
图12是实施例13产物1-烯丙基-3,4-二氢异喹啉的核磁氢碳谱图;
[0040]
图13是实施例14产物6,7-二甲氧基-1-(2-甲基烯丙基)-3,4-二氢异喹啉的核磁氢碳谱图;
[0041]
图14是实施例15产物6-溴-1-(2-甲基烯丙基)-3,4-二氢异喹啉的核磁氢碳谱图。
具体实施方式
[0042]
结合以下具体实施例,对本发明作进一步的详细说明,本发明的保护内容不局限于以下实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。实施本发明的过程、条件、试剂、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。以下实施例所给出的数据包括具体操作和反应条件及产物。产物纯度通过核磁、高分辨质谱鉴定。
[0043]
实施例1
[0044]
(e)-2-(对甲苯基二氮烯基)-1,2,3,4-四氢异喹啉的合成
[0045][0046]
在100ml的三口烧瓶中,加入对甲基苯胺(10mmol),然后在0℃下将2ml的质量浓度12.0mol/l浓盐酸入三口烧瓶中。在50ml的烧杯中加入亚硝酸钠0.76g(11mmol),再加入5ml
的蒸馏水使其溶解,然后用恒压滴液漏斗将亚硝酸钠水溶液逐滴加入到对甲基苯胺的盐酸溶液中,在0℃下继续搅拌10min,得对甲基苯基重氮盐溶液;
[0047]
在50ml烧杯中加入四氢异喹啉(10mmol)和1.2mol/l的碳酸钾溶液(1.6585g溶于10ml水),搅拌均匀后一次性将上述对甲基苯基重氮盐溶液加入其中,在0℃搅拌反应半小时后出现大量黄色固体,用布氏漏斗抽滤并用冰水洗涤固体,得粗产品,将无水乙醇加热到60℃,将粗产品转移至圆底烧瓶中,滴加60℃无水乙醇重结晶,得到淡黄色固体产物,产率(2.26g,90%)。该化合物经核磁、高分辨质谱表征,该化合物核磁、高分辨质谱数据与文献报道的数据一致(n.jung,s.brase.sci.synth.2010,41,613-640.)。
[0048]
实施例2
[0049]
(e)-6,7-二甲氧基-2-(对甲苯基二氮烯基)-1,2,3,4-四氢异喹啉的合成
[0050][0051]
在100ml的三口烧瓶中,加入对甲基苯胺(10mmol),然后在0℃下将2ml的质量浓度12.0mol/l浓盐酸入三口烧瓶中。在50ml的烧杯中加入亚硝酸钠0.76g(11mmol),再加入5ml的蒸馏水使其溶解,然后用恒压滴液漏斗将亚硝酸钠水溶液逐滴加入到对甲基苯胺的盐酸溶液中,在0℃下继续搅拌10min,得对甲基苯基重氮盐溶液;
[0052]
在50ml烧杯中加入6,7-二甲氧基四氢异喹啉(10mmol)和1.2mol/l的碳酸钾溶液(1.6585g溶于10ml水),搅拌均匀后一次性将上述对甲基苯基重氮盐溶液加入其中,在0℃搅拌反应半小时后出现大量黄色固体,用布氏漏斗抽滤并用冰水洗涤固体,得粗产品,将无水乙醇加热到60℃,将粗产品转移至圆底烧瓶中,滴加60℃无水乙醇重结晶,得到淡黄色固体产物,产率(2.12g,88%),熔点为:137-138℃。该化合物经核磁、高分辨质谱表征,所得产品的参数为1h nmr(400mhz,cdcl3)δ7.29(d,j=8.2hz,2h),7.08(d,j=8.2hz,2h),6.62(d,j=14.7hz,2h),4.78(s,2h),3.99(t,j=5.9hz,2h),3.80(s,3h),3.79(s,3h),2.91(t,j=5.9hz,2h),2.27(s,3h)ppm.
13
c nmr(100mhz,cdcl3)δ148.68,148.05,147.92,135.56,129.62,128.27,126.69,120.56,111.46,109.81,56.11,48.33,47.67,29.18,21.17ppm.hrms(esi)m/z calcd for 2.c
18h21
n3o2[m h]
:312.17065;found:312.16977.
[0053]
实施例3
[0054]
(e)-6-溴-2-(对甲苯基二氮烯基)-1,2,3,4-四氢异喹啉的合成
[0055][0056]
在100ml的三口烧瓶中,加入对甲基苯胺(10mmol),然后在0℃下将2ml的质量浓度12.0mol/l浓盐酸入三口烧瓶中。在50ml的烧杯中加入亚硝酸钠0.76g(11mmol),再加入5ml的蒸馏水使其溶解,然后用恒压滴液漏斗将亚硝酸钠水溶液逐滴加入到对甲基苯胺的盐酸溶液中,在0℃下继续搅拌10min,得对甲基苯基重氮盐溶液;
[0057]
在50ml烧杯中加入6-溴四氢异喹啉(10mmol)和1.2mol/l的碳酸钾溶液(1.6585g溶于10ml水),搅拌均匀后一次性将上述对甲基苯基重氮盐溶液加入其中,在0℃搅拌反应半小时后出现大量黄色固体,用布氏漏斗抽滤并用冰水洗涤固体,得粗产品,将无水乙醇加热到60℃,将粗产品转移至圆底烧瓶中,滴加60℃无水乙醇重结晶,得到淡黄色固体产物,产率(2.12g,88%),熔点为:108-109℃。该化合物经核磁、高分辨质谱表征,所得产品的参数为1h nmr(400mhz,cdcl3)δ7.36(d,j=8.3hz,3h),7.31(d,j=8.1hz,1h),7.15(d,j=8.4hz,2h),7.04(d,j=8.1hz,1h),4.88(s,2h),4.05(t,j=5.9hz,2h),2.98(t,j=5.9hz,2h),2.34(s,3h)ppm.
13
c nmr(100mhz,cdcl3)δ148.43,135.87,133.90,132.40,130.28,129.95,129.89,129.66,129.18,128.26,120.64,120.28,49.03,47.82,29.14,21.20ppm.hrms(esi)m/z calcd for 3.c
16h16
brn3[m h]
:330.06004;found:330.05939.
[0058]
实施例4
[0059]
1-(2-甲烯丙基)-3,4-二氢异喹啉的合成
[0060][0061]
在干燥清洁的石英管中加入实施例1制备的反应底物(i)(0.2mmol,50.2mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,54ul),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(23.32mg,收率63%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.10(dd,j=7.7,1.2hz,1h),7.41(td,j=7.4,1.5hz,1h),7.34(t,j=6.9hz,1h),7.18(d,j=7.5hz,1h),4.91(d,j=11.9hz,2h),4.16(s,2h),3.48(t,j=6.6hz,2h),2.98(t,j=6.6hz,2h)ppm.
13
c nmr(100mhz,cdcl3)δ164.50,141.08,138.20,131.74,129.58,128.55,127.16,126.99,52.70,45.11,28.23,20.19ppm.hrms(esi)m/z calcd for c
13h15
n[m h]
:186.12773;found:186.12746.说明在上述条件作用下时,可获得最高产率的目标产物。
[0062]
实施例5
[0063]
1-苄基-3,4-二氢异喹啉的合成反应
[0064][0065]
在干燥清洁的石英管中加入实施例1制备的反应底物(i)(0.2mmol,50.2mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,48ul),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(23.44mg,收率55%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.15(d,j=7.4hz,1h),7.44-7.39(m,1h),7.38-7.27(m,6h),7.16(d,j=7.1hz,1h),7.28(d,j=3.9hz,1h),7.16(d,j=7.1hz,1h),4.80(s,2h),3.48(t,j=6.4hz,2h),2.93(t,j=6.4hz,2h)ppm.
13
c nmr(100mhz,cdcl3)δ164.69,138.17,137.57,131.81,129.50,129.50,128.75,128.57,128.17,127.56,127.18,127.02,50.56,45.47,28.21ppm.hrms(esi)m/z calcd for c
16h15
n[m h]
:222.12773;found:222.12685.
[0066]
化合物iii(0.2mmol,44.2mg)经过在(1,5-环辛二烯)氯化铱(i)二聚体(0.5mol%,0.7mg)和(ra,ss)-3a(2.2mol%,2.2mg)催化下,碘化钾(10mol%,3.3mg),氢气(1atm)作为还原剂,以叔丁基甲基醚(2ml)作为溶剂,常温反应顺利转化为1-苯基-1,2,3,4-四氢异喹啉(收率:96%,42.8mg)(采用核磁共振仪、高分辨质谱检测),因具有药理活性而被作为合成药物分子的核心骨架(j.-h.xie,p.-c.yan,q.-q.zhang,k.-x.yuan,q.-l.zhou.acs catal.2012,2,561-564.)。
[0067]
实施例6
[0068]
1-(4-甲基苄基)-3,4-二氢异喹啉的合成反应
[0069]
[0070]
在干燥清洁的石英管中加入实施例1制备的反应底物(i)(0.2mmol,50.2mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,74.1mg),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(24.92mg,收率53%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.14(d,j=7.4hz,1h),7.40(dt,j=7.4,3.7hz,1h),7.35(t,j=7.3hz,1h),7.22(d,j=7.9hz,2h),7.14(t,j=6.9hz,3h),4.75(s,2h),3.46(t,j=6.6hz,2h),2.91(t,j=6.6hz,2h),2.33(s,3h)ppm.
13
c nmr(100mhz,cdcl3)δ164.64,138.17,137.21,134.50,131.76,129.55,129.41,128.55,128.19,127.15,126.99,50.24,45.32,28.21,21.22ppm.hrms(esi)m/z calcd for c
17h11
n[m h]
:236.14338;found:236.14314.
[0071]
实施例7
[0072]
1-(3-甲基苄基)-3,4-二氢异喹啉的合成反应
[0073][0074]
在干燥清洁的石英管中加入实施例1制备的反应底物(i)(0.2mmol,50.2mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,54ul),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(24.92mg,收率53%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.15(d,j=7.4hz,1h),7.42(td,j=7.3,1.0hz,1h),7.35(t,j=7.3hz,1h),7.21(t,j=7.5hz,1h),7.12(dt,j=15.8,7.3hz,4h),4.76(s,2h),3.48(t,j=6.6hz,2h),2.93(t,j=6.6hz,2h),2.33(s,3h)ppm.
13
c nmr(100mhz,cdcl3)δ164.64,138.45,138.19,137.48,131.78,129.54,128.87,128.60,128.56,128.32,127.16,127.01,125.23,50.46,45.39,28.20,21.50ppm.hrms(esi)m/z calcd for c
17h17
n[m h]
:236.14338;found:236.14311.
[0075]
实施例8
[0076]
1-(2-甲基苄基)-3,4-二氢异喹啉的合成反应
[0077][0078]
在干燥清洁的石英管中加入实施例1制备的反应底物(i)(0.2mmol,50.2mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,54ul),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(25.39mg,收率54%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.15(d,j=7.7hz,1h),7.43(td,j=7.4,1.4hz,1h),7.36(t,j=7.2hz,1h),7.24-7.20(m,2h),7.20-7.15(m,3h),4.82(s,2h),3.44(t,j=6.6hz,2h),2.93(t,j=6.6hz,2h),2.33(s,3h)ppm.
13
c nmr(100mhz,cdcl3)δ164.58,138.15,136.93,135.01,131.83,130.71,129.51,128.62,127.65,127.21,127.02,126.18,48.32,44.98,28.17,19.41ppm.hrms(esi)m/z calcd for c
17h17
n[m h]
:236.14338;found:236.14305.
[0079]
实施例9
[0080]
1-(4-异丙基苄基)-3,4-二氢异喹啉的合成反应
[0081][0082]
在干燥清洁的石英管中加入实施例1制备的反应底物(i)(0.2mmol,50.2mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,67ul),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(22.11mg,收率42%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.14(dd,j=7.6,1.0hz,1h),7.41(td,j=7.4,1.4hz,1h),7.35(t,j=7.3hz,1h),7.25(d,j=2.1hz,2h),7.18(d,j=8.1hz,2h),7.15(d,j=7.4hz,1h),4.76(s,2h),3.48(t,j=6.6hz,2h),2.93(t,j=6.6hz,2h),2.88(dd,j=13.8,6.9hz,1h),1.23(d,j=6.9hz,6h)ppm.
13
c nmr(100mhz,cdcl3)δ164.64,148.21,138.19,134.85,131.75,129.58,128.55,128.18,127.14,126.99,126.78,50.32,45.45,33.90,28.22,24.10ppm.hrms(esi)m/z calcd for c
19h21
n[m h]
:264.17468;found:264.17435.
[0083]
实施例10
[0084]
1-(4-氯苄基)-3,4-二氢异喹啉的合成反应
[0085][0086]
在干燥清洁的石英管中加入实施例1制备的反应底物(i)(0.2mmol,50.2mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,54ul),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(21.43mg,收率42%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.14(d,j=7.5hz,1h),7.43(t,j=7.0hz,1h),7.36(t,j=7.4hz,1h),7.29(d,j=2.2hz,4h),7.16(d,j=7.3hz,1h),4.75(s,2h),3.48(t,j=6.6hz,2h),2.94(t,j=6.6hz,2h)ppm.
13
c nmr(100mhz,cdcl3)δ164.73,138.11,136.15,133.41,131.96,129.56,129.32,128.92,128.58,127.25,127.08,50.03,45.59,28.20ppm.hrms(esi)m/z calcd for c
16h14
ncl[m h]
:256.08875;found:256.08829.
[0087]
实施例11
[0088]
1-(4-溴苄基)-3,4-二氢异喹啉的合成反应
[0089][0090]
在干燥清洁的石英管中加入实施例1制备的反应底物(i)(0.2mmol,50.2mg)、k2co3(0.25mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,99.97mg),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中
加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(29.31mg,收率49%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.13(dd,j=7.6,0.9hz,1h),7.45(d,j=8.3hz,2h),7.42(dd,j=7.5,1.4hz,1h),7.36(t,j=7.3hz,1h),7.21(d,j=8.3hz,2h),7.16(d,j=7.4hz,1h),4.73(s,2h),3.47(t,j=6.6hz,2h),2.94(t,j=6.6hz,2h)ppm.
13
c nmr(100mhz,cdcl3)δ164.75,138.10,136.65,131.97,131.87,129.90,129.27,128.57,127.25,127.08,121.49,50.08,45.59,28.18ppm.hrms(esi)m/z calcd for c
16h14
nbr[m h]
:300.03824;found:300.03827.
[0091]
化合物iii(0.2mmol,59.8mg)经过在(1,5-环辛二烯)氯化铱(i)二聚体(0.5mol%,0.7mg)和(ra,ss)-3a(2.2mol%,2.2mg)催化下,碘化钾(10mol%,3.3mg),氢气(1atm)作为还原剂,以叔丁基甲基醚(2ml)作为溶剂,常温反应顺利转化为1-苯基-1,2,3,4-四氢异喹啉(收率:94%,56.6mg)(采用核磁共振仪、高分辨质谱检测),因具有药理活性而被作为合成药物分子的核心骨架(j.-h.xie,p.-c.yan,q.-q.zhang,k.-x.yuan,q.-l.zhou.acs catal.2012,2,561-564.)。
[0092]
实施例12
[0093]
1-(1-苯乙基)-3,4-二氢异喹啉的合成反应
[0094][0095]
在干燥清洁的石英管中加入实施例1制备的反应底物(i)(0.2mmol,50.2mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,55ul),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(23.51mg,收率50%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.14(dt,j=20.4,10.2hz,1h),7.39(dt,j=6.8,2.5hz,3h),7.37-7.32(m,3h),7.28(dt,j=5.1,2.0hz,1h),7.13(dd,j=7.3,0.6hz,1h),6.25(q,j=7.1hz,1h),3.39(ddd,j=12.4,8.0,6.4hz,1h),3.23-3.01(m,1h),2.82(t,j=6.5hz,1h),1.59(d,j=7.1hz,3h)ppm.
13
c nmr(100mhz,cdcl3)δ164.36,140.93,138.14,131.75,129.87,128.67,128.59,127.46,127.44,127.18,126.93,50.35,40.24,28.46,15.81ppm.hrms(esi)m/z calcd for c
17h17
n[m h]
:236.14338;found:236.14304.
[0096]
实施例13
[0097]
1-烯丙基-3,4-二氢异喹啉的合成反应
[0098][0099]
在干燥清洁的石英管中加入实施例1制备的反应底物(i)(0.2mmol,50.2mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.6mmol,3.0equiv.,72ul),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(15.55mg,收率42%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.03(d,j=7.5hz,1h),7.34(td,j=7.5,1.1hz,1h),7.27(t,j=7.4hz,1h),7.10(d,j=7.4hz,1h),5.79(ddt,j=16.1,10.2,5.9hz,1h),5.27-5.01(m,2h),4.14(d,j=5.9hz,2h),3.45(t,j=6.6hz,2h),2.92(t,j=6.6hz,2h)ppm.
13
c nmr(100mhz,cdcl3)δ164.39,138.20,133.30,131.75,129.58,128.48,127.16,127.01,117.57,49.73,45.46,28.23ppm.hrms(esi)m/z calcd for c
12h13
n[m h]
:172.11208;found:172.11187.
[0100]
实施例14
[0101]
6,7-二甲氧基-1-(2-甲基烯丙基)-3,4-二氢异喹啉的合成反应
[0102]
在干燥清洁的石英管中加入实施例2制备的反应底物(i)(0.2mmol,62.23mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,54ul),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(19.62mg,收率40%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ7.55(s,1h),6.57(s,1h),4.83(d,j=11.2hz,2h),4.06(s,2h),3.85(s,2h),3.84(s,2h),3.40(t,j=6.7hz,2h),2.84(t,j=6.7hz,2h),1.67(s,3h)ppm.
13
c nmr(100mhz,cdcl3)δ164.63,151.97,148.17,141.36,131.88,122.20,112.81,110.91,109.45,56.27,56.23,52.78,45.44,27.92,20.26ppm.hrms(esi)m/z calcd for c
15h19
no2[m h]
:246.14886;found:246.14926.
[0103]
实施例15
[0104]
6-溴-1-(4-甲基苄基)-3,4-二氢异喹啉的合成反应
[0105][0106]
在干燥清洁的石英管中加入实施例3制备的反应底物(i)(0.2mmol,65.81mg)、k2co3(0.5mmol,2.5equiv.,69.1mg)和18-crown-6(0.3mmol,1.5equiv.,79.3mg),然后,密闭封管管口在脱气充入n2(1atm),在氮气氛围下加入溶剂ch3cn(2ml)和化合物(ii)(0.4mmol,2.0equiv.,74.1mg),最后在紫光(390-395nm)照射下反应36h。反应完成后,向反应体系中加入5ml的水淬灭反应,用乙酸乙酯萃取(3
×
5ml,共进行3次,每次5ml)含水混合物。萃取完后有机相用无水硫酸镁干燥,过滤并减压浓缩,通过柱色谱(洗脱剂:石油醚/乙酸乙酯15:1-5:1,v/v,(在此为5:1))分离纯化得到黄色固体(26.30mg,收率42%)。采用核磁共振仪、高分辨质谱检测,所得产品的参数为1h nmr(400mhz,cdcl3)δ8.19(s,1h),7.44(d,j=7.9hz,1h),7.13(d,j=7.5hz,2h),7.05(d,j=7.3hz,2h),6.95(d,j=7.9hz,1h),4.65(s,2h),3.37(t,j=6.1hz,2h),2.78(t,j=6.1hz,2h),2.25(s,3h).
13
c nmr(100mhz,cdcl3)δ163.33,137.41,136.92,134.61,134.17,131.52,129.49,128.76,128.22,50.38,45.14,27.72,21.24.hrms(esi)m/z calcd for c
17h16
brn[m h]
:314.05389;found:314.05469.
[0107]
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
