1.本发明涉及食品安全领域,具体的说涉及一种高效制备交链孢酚的方法。
背景技术:
2.交链孢酚(alternariol,aoh)是由链格孢属(alternaria spp.)真菌侵染农作物后在适宜温度和湿度下产生的一种二苯并-α-吡喃酮类链格孢霉毒素。它在1953年首次由raistrick从微生物中分离得到,为白色粉末状固体,分子式:c
14h10
o5,分子量:258.23,易溶于甲醇、乙醇、乙腈和二甲基亚砜等有机溶剂。
3.交链孢酚是在谷物、饲料及果蔬中存在的最普遍的链格孢霉毒素之一,可通过食物链对人类和动物健康造成严重危害。研究表明,交链孢酚主要通过造成机体的氧化应激产生ros并与dna拓扑异构酶相互作用,导致dna单链和双链断裂,进而阻止细胞增殖,此外,交链孢酚在结构上类似于雌二醇,表现出雌激素效应并干扰类固醇生成。
4.欧洲食品安全局(efsa)采用毒理学关注阈值(ttc)来评价交链孢酚对人体造成的潜在风险,其ttc值为2.5μg/kg body weight/day。但是目前尚无适用于食品和饲料中交链孢酚的限量标准,且交链孢酚的代谢分布研究较少,其进入动物体后的动力学规律仍不清楚,而研究aoh的毒理学及其在体内的迁移转化、分析检测和风险评估等需要用到大量的标准品。
5.目前,交链孢酚的制备方法主要为溶剂分配法、索氏抽提法和硅胶柱层析法等,但这些方法都很耗时且目标物纯度较低,不适用于大规模制备。chu等以大米培养基为产毒基质,二氯甲烷为提取溶剂,首次采用半制备及制备液相分离纯化三种链格孢霉毒素,极大地提高了制备效率且目标物纯度达到95%,但该方法培养周期较长,提取溶剂毒性较大,制备步骤复杂,不适用于工业级制备。而且目前的交链孢酚标准品,价格昂贵约为1500元/mg。
6.因此,迫切需要开发新的交链孢酚的纯化制备方法。
技术实现要素:
7.本发明的目的在于提供一种高效制备交链孢酚的方法,该方法包括如下步骤:将互隔交链孢菌接种在pda培养基上,黑暗条件下活化培养;取小块接种于新的培养基进行培养;在旋转涡旋仪上涡旋,收集提取液;提取液过滤、蒸干;乙酸乙酯提取、蒸干;两次中压制备;重结晶。
8.具体的说,本发明的一种高效制备交链孢酚的方法,该方法包括如下步骤:
9.(1)将互隔交链孢菌(atcc 66981,购自美国模式培养物集存库(american typeculture collection,atcc))接种在pda培养基上,20-40℃黑暗条件下活化培养3-10d;
10.(2)用无菌牙签从步骤(1)得到的pda培养基菌丝边缘上取小块接种于新的培养基进行培养,培养的条件为:
11.③
pda或pdb培养基(培养基初始ph 4-7)在20-40℃黑暗条件下培养5-10d;或者
是:
12.④
大米或玉米培养基在20-40℃黑暗条件下培养20-40d;
13.(3)将步骤(2)得到的培养基转移到锥形瓶中,加入乙腈,在旋转涡旋仪以100-500rpm/min的速度涡旋1-5min,然后超声0.5-3h,重复此步骤2-5次,收集提取液;
14.(4)将步骤(3)得到的提取液用纱布过滤到旋蒸瓶中,旋转蒸发仪30-60℃下旋蒸,直至液体全部蒸干;
15.(5)将蒸干后的固体用纯水复溶转移到分液漏斗中,加入乙酸乙酯(乙酸乙酯与水的比例为1:0.5-3(v/v)),剧烈摇晃20-100次左右,静置等待液体分层,收集上层溶液,重复此步骤2-5次,将萃取后收集的液体再次旋蒸浓缩至干,得到固体物质;
16.(6)将步骤(5)收集的固体物质经甲醇/乙腈或其它有机溶剂溶解后过0.22μm滤膜,使用中压制备液相仪,依次进行两次中压制备:
17.一次制备条件:unitary c
18
制备色谱柱,10mm
×
250mm,5μm或其它等效柱;流动相a为0.1-1%的甲酸-水或者0.1-1%的乙酸水,b为甲醇或乙腈(或其它有机相),流速为5-30ml/min,检测波长为256nm,进样量为1ml,收集10~25min馏分液,将馏分液30-60℃旋蒸浓缩至干,再次复溶后进行二次制备;
18.二次制备条件:unitary c
18
制备色谱柱,10mm
×
250mm,5μm或其它等效柱;流动相a为0.1-1%的甲酸-水或者0.1-1%的乙酸水,b为甲醇或乙腈(或其它有机相),流速为5-30ml/min,检测波长为256nm,进样量为1ml,收集10~25min馏分液,30-60℃水浴旋蒸后置于冷冻真空干燥机干燥,获得淡黄色固体;
19.(7)将(6)获得的淡黄色固体通过甲醇、乙腈、丙酮或其它有机溶剂进行重结晶,吸去上清液,重复3-20次,直到上清液澄清透明得到结晶后的交链孢酚。
20.更为具体的说,本发明的一种高效制备交链孢酚的方法,包括如下步骤:
21.(1)将互隔交链孢菌接种在pda培养基上,28℃黑暗条件下活化培养5d;
22.(2)用无菌牙签从步骤(1)得到的pda培养基菌丝边缘上取小块接种于新的培养基进行培养,培养的条件为:
23.pda培养基,培养基初始ph 6.4,在28℃黑暗条件下培养8d;
24.(3)将步骤(2)得到的培养基转移到锥形瓶中,加入乙腈,在旋转涡旋仪以200rpm/min的速度涡旋2min,然后超声1h,重复此步骤3次,收集提取液;
25.(4)将步骤(3)得到的提取液用纱布过滤到旋蒸瓶中,旋转蒸发仪50℃下旋蒸,直至液体全部蒸干;
26.(5)将蒸干后的固体用纯水复溶转移到分液漏斗中,加入等体积的乙酸乙酯,剧烈摇晃,静置等待液体分层,收集上层溶液,重复此步骤2-5次,将萃取后收集的液体再次旋蒸浓缩至干,得到固体物质;
27.(6)将步骤(5)收集的固体物质经甲醇或乙腈溶解后过0.22μm滤膜,使用中压制备液相仪,依次进行两次中压制备,获得淡黄色固体;
28.一次制备条件:unitary c
18
制备色谱柱,10mm
×
250mm,5μm;流动相a为0.1%的甲酸-水,b为甲醇,流速为20ml/min,检测波长为256nm,进样量为1ml,收集15~17.5min馏分液,将馏分液50℃旋蒸浓缩至干,再次复溶后进行二次制备;
29.二次制备条件:unitary c
18
制备色谱柱,10mm
×
250mm,5μm;流动相a为0.1%的甲
酸-水,b为甲醇,流速为20ml/min,检测波长为256nm,进样量为1ml,收集18.4~19.7min馏分液,50℃水浴旋蒸后置于冷冻真空干燥机干燥,获得淡黄色固体;
30.(7)将(6)获得的淡黄色固体通过甲醇、乙腈或丙酮进行重结晶,吸去上清液,重复2-5次,直到上清液澄清透明得到结晶后的交链孢酚。
31.将制备得到的交链孢酚通过超高效液相色谱(uplc-pda)面积归一法和定量核磁检测其纯度。
32.本发明提供了一种简单快速、价格低廉、纯度高且可大量制备交链孢酚的方法。该方法选择产毒量高、杂质较少和培养时间短的pda培养基为产毒培养基,然后经过提取、萃取后通过制备液相分离纯化及重结晶,最终获得了纯度较高的目标化合物。该工艺操作简单、制备效率快,可大幅度降低生产成本,同时纯度达到99%以上,可作为标准品应用于食品中交链孢酚的检测、防控及其毒理学研究。
33.与现有技术相比,本发明有以下有益的技术效果:
34.(1)互隔交链孢菌atcc 66981在最佳培养条件下得到交链孢酚的产量高达2096.1mg/kg;
35.(2)高产交链孢酚的培养基为马铃薯葡萄糖培养基(pda培养基)时,培养时间短;
36.(3)获得的aoh高含量培养基杂质含量少,简化提取净化步骤,保护色谱柱,极大的提高了制备效率,显著降低成本。
37.(4)采用反相制备色谱法结合重结晶法纯化得到高纯度的交链孢酚,显著提升了标准物质的纯度的同时提高了制备效率。
附图说明
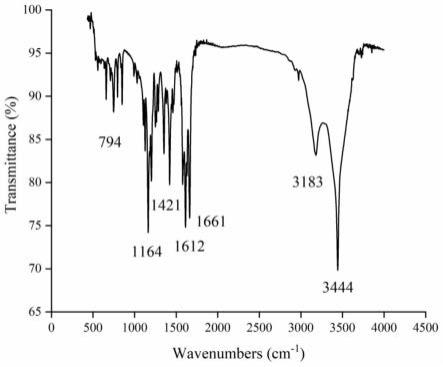
38.图1为制备品aoh的红外光谱图
39.图2为制备品aoh的核磁氢谱
40.图3为制备品aoh的碳谱
41.图4为制备品aoh的紫外吸收波谱图
42.图5为esi
和esi-模式下制备品aoh的飞行时间质谱图
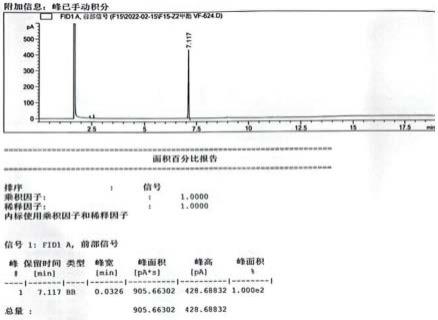
43.图6为aoh收集液的液相色谱图
具体实施方式
44.实施例一:
45.下列实施例中所用的pda培养基为:4g pda粉末加100ml水溶解而成(pda粉末:北京陆桥技术有限责任公司)
46.1、产毒培养
47.将互隔交链孢菌株(atcc 66981,购自美国模式培养物集存库(american type culture collection,atcc))接种在pda培养基上,28℃黑暗条件下活化培养5d,用无菌牙签从培养基边缘上取小块接种于新的pda培养基中,培养基初始ph 6.4,培养条件为培养温度28.0℃、培养时间8d。
48.2、培养基中aoh含量测定
49.培养基通过uplc xevotq-s超高效液相色谱-串联质谱联用仪(美国waters公司)
进行测定,交链孢酚的产量高达2096.1mg/kg。
50.3、样品提取
51.将上述pda培养基转移到1l的锥形瓶中,加入900ml乙腈,在旋转涡旋仪以200rpm/min的速度涡旋2min,然后超声1h,重复此步骤三次。将提取液用四层纱布(100目)过滤到旋蒸瓶中,50℃旋转蒸发仪浓缩,直至液体全部蒸干。
52.4、萃取
53.将旋蒸干的固体用水复溶(溶解至无物体颗粒)转移到分液漏斗中,加入等体积的乙酸乙酯,剧烈摇晃50次左右,静置等待液体分层,将上清液(乙酸乙酯层)收集,重复此步骤3次,将萃取后的液体再次旋蒸浓缩至干,得到固体物质。
54.5、将上述固体物质进行两次中压制备
55.5.1一次制备
56.色谱条件:unitary c
18
制备色谱柱(10mm
×
250mm,5μm,浙江华谱新创科技有限公司);流动相a为0.1%(v/v)甲酸-水,b为甲醇;流速20ml/min;进样量1000μl。洗脱梯度见表1。
57.表1一次制备梯度洗脱条件
[0058][0059]
馏分采集条件:经unitary c
18
制备色谱柱分离使杂质和目标物aoh完全分离,在15~17.5min时间段(表2)收集aoh馏分,防止杂质和目标物交叉污染。将馏分液50℃旋蒸浓缩至干,再次复溶后进行二次制备。
[0060]
表2 aoh馏分收集时间
[0061][0062]
5.2二次制备条件
[0063]
色谱条件:unitary c
18
制备色谱柱(10mm
×
250mm,5μm,浙江华谱新创科技有限公司);流动相a为0.1%(v/v)甲酸-水,b为甲醇;流速20ml/min;进样量1000μl。洗脱梯度见表3。
[0064]
表3二次制备梯度洗脱条件
[0065][0066]
馏分采集条件:经unitary c
18
制备色谱柱二次制备使杂质和目标物aoh进一步分离,在固定时间段(18.4~19.7min)收集aoh馏分(表4),防止杂质和目标物交叉污染。收集馏分液,50℃水浴旋蒸后置于冷冻真空干燥机干燥,获得淡黄色固体。
[0067]
表4aoh馏分收集时间
[0068][0069]
6、重结晶
[0070]
将获得的淡黄色固体通过甲醇重结晶后得到结晶后的交链孢酚。
[0071]
7、冷冻干燥
[0072]
将获得的结晶后的aoh固体进一步置于-80℃保存过夜后置冷冻干燥机干燥,直至完全冻干,将固体粉末取出,-20℃保存。
[0073]
实施例二
[0074]
对实施例一制备得到的aoh进行定性分析及纯度测定
[0075]
1、红外光谱鉴定
[0076]
红外光谱是表征化合物主要组成基团的有力手段。实施例一制备的aoh的ft-ir光谱如图1所示,ft-ir光谱中出现了几个主要吸收带,分别是在3444、3183、1661、1612、1421、1164和794cm-1
,表明实施例一所制备的化合物为aoh。
[0077]
2、核磁共振鉴定
[0078]
nmr是分析与鉴定化合物结构的一种较为有效的手段,常常被用于制备样品鉴定及纯度测定。将实施例一中制备得到的aoh固体粉末溶于dmso-d6(纯度99.9%,上海阿达玛斯试剂有限公司),1h和
13
c核磁图谱分别如图2和图3所示,进一步证明制备品为aoh。
[0079]
3、紫外鉴定
[0080]
如图4所示,制备品在255.7nm、299.4nm和338.9nm有紫外吸收,进一步证明实施例一的制备品为aoh。
[0081]
4、飞行时间质谱鉴定
[0082]
lc-q-tof/ms条件:acquity beh c
18
色谱柱(2.1mm
×
100mm,1.7μm,美国waters公司);流动相a为乙腈,b为水(含有3mmol/l乙酸铵和0.1%v/v的甲酸),梯度洗脱:0~2min,5%b~15%b;2~10min,15%b~60%b;10~15min,60%b~90%b;15~16min,90%b~90%b;16~16.5min,90%b~5%b;16.5~18min,5%b~5%b。流速0.4ml/min,进样量为8μ
l。雾化气压采用电喷雾esi离子源,离子源温度550℃,喷雾电压3.5kv(esi
)和5.5kv(esi
–
);去簇电位和碰撞能量分别为80v和10v;扫描范围100~900(m/z);碎裂电压为1.5v,扫描力3500pa;鞘气压45pa;辅助气压15pa。
[0083]
将实施例一获得的aoh在正负离子模式进行全扫描,均得到单一且高峰度的色谱峰。然后进一步对特征峰在正离子模式和负离子模式下进行母离子扫描,在正离子模式下,化合物的主要分子离子峰为259.04[m h]
,主要分子离子峰的主要二级碎片213.04、185.04、128.05;在负离子模式下,化合物的主要分子离子峰为257.04[m-h]-,主要分子离子峰的主要二级碎片213.05、212.04、147.04,进一步证明制备品为aoh(图5)。
[0084]
5、超高效液相色谱仪配二极管阵列检测器(uplc-pda)全扫描
[0085]
色谱条件:acquity beh c
18
色谱柱(2.1mm
×
100mm,1.7μm,美国waters公司);流动相a为乙腈,b为纯水;0~8min,40%a~60%a;8.0~8.5min,60%a~90%a;8.5~10.5min,90%a~90%a;10.5~11.0min,90%a~40%a;11~15min,40%a~40%a;流速0.3ml/min;进样量5μl;紫外检测器;波长210nm~400nm。
[0086]
通过uplc-pda全扫描制备得到的aoh制备品进行纯度分析,结果发现通过面积归一法测得aoh的纯度达到99%以上(图6)。
[0087]
6、定量核磁测定纯度
[0088]
称取10mg实施例一制备得到的aoh和10mg苯甲酸(国家标准物质gbw06117,纯度99.99%,中国计量科学研究院)于核磁管中,然后加入550μl dmso-d6(纯度99.9%,上海阿达玛斯试剂有限公司)充分溶解,将核磁管进行核磁共振仪(ac-80,bruker biospin gmbh,德国)测定,仪器频率为500hz。利用mestrenova 14软件(西班牙mestrelab research公司)进行数据处理,得到aoh的纯度为99.3%。
[0089]
综上所述,实施例一的高效制备交链孢酚的方法,培养周期仅为8天且pda培养基提取液中杂质较少,保护色谱柱,提高了制备效率,可大幅度降低生产成本,同时纯度达到99%以上,可作为标准品应用于食品中交链孢酚检测、防控及其毒理学研究。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。