1.本发明涉及化学合成技术领域,尤其是指一种吡咯啉酮类化合物的合成方法。
背景技术:
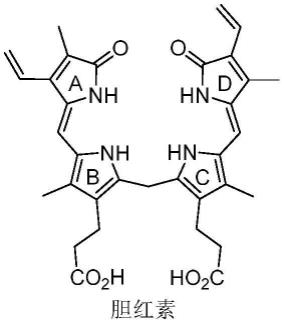
2.吡咯啉酮是胆红素(结构式如下所示)、胆绿素、藻蓝素等药物和食品添加剂的重要结构片段,胆红素中,a环和d环均为吡咯啉酮。
[0003][0004]
公开号为cn112592306a的中国发明专利公开了一种吡咯啉酮类化合物及其合成方法,该专利是胆红素d环的合成方法。现有技术中,有关胆色素(包括胆红素)a环的合成方法文献报道路线很少。其中,德国化学家plieninger用10步合成了乙烯基a环【liebigs ann.chem.723,149-158(1969)】,gossauer用氰化钠在加压条件下合成了a环前体(3-甲基-4-乙酸基吡咯啉酮),然而,收率只有10%【helveticcah imicacta-vol.72,518(1989)】。胆红素是牛黄(包括天然牛黄、体外培育牛黄)的主要有效成分,a环是合成胆色素(包括胆红素)的关键中间体。
[0005]
因此,研发一种较短的合成路线制备吡咯啉酮,特别是制备4-乙烯基吡咯啉酮(a环),具有较大的理论和工业价值。
技术实现要素:
[0006]
本发明旨在至少在一定程度上解决现有技术中存在的技术问题之一,在本发明的第一方面,本发明提供一种吡咯啉酮类化合物,所述吡咯啉酮类化合物的结构式如式1所示,
[0007][0008]
其中,r1为c1~c12烷基;r2取自氢、苄基、取代苄基中的一种;优选地,r1为乙烯基或α对甲基苯磺酰基乙基,r2为氢或对甲氧基苄基或苄基。
[0009]
在本发明的第二方面,本发明提供一种吡咯啉酮类化合物中间体,所述吡咯啉酮类化合物中间体的结构式如式6所示,
[0010][0011]
其中,r2取自氢、苄基、取代苄基中的一种,r3取自乙烯基或ch3ch,r4取自羟基取代甲基或醛基;优选地,r2为对甲氧基苄基或苄基。
[0012]
在本发明的第三方面,本发明提供一种在本发明第一方面所述的吡咯啉酮类化合物的制备方法,所述的吡咯啉酮类化合物如式1a所示,式1a所示化合物由式1b所示化合物制备得到,式1b所示化合物制备式1a所示化合物的反应式如下所示:
[0013][0014]
其中,r2取自苄基、取代苄基中的一种;优选地,r2为对甲氧基苄基或苄基。
[0015]
在本发明的第四方面,本发明提供一种在本发明第一方面所述的另一种吡咯啉酮类化合物的制备方法,其特征在于,式1所示化合物由式4所示化合物制备得到,式4所示化合物制备
[0016]
式1所示化合物的反应式如下所示:
[0017][0018]
其中,r2取自苄基、取代苄基中的一种,r1为c1~c12烷基;优选地,r2为对甲氧基苄基或苄基,r1为乙烯基或α对甲基苯磺酰基乙基。
[0019]
在本发明的一个或多个实施例中,所述式4所示化合物由式3所示化合物制备得到,式3所示化合物制备式4所示化合物的反应式如下所示:
[0020][0021]
在本发明的一个或多个实施例中,所述式3所示化合物由式2所示化合物制备得到,式2所示化合物制备式3所示化合物的反应式如下所示:
[0022][0023]
在本发明的一个或多个实施例中,所述式2所示化合物由式5所示化合物制备得到,式5所示化合物制备式2所示化合物的反应式如下所示:
[0024][0025]
在本发明的第五方面,本发明提供一种在本发明第二方面所述的吡咯啉酮类化合物中间体的制备方法,当r3为ch3ch,r4为醛基时,即所述吡咯啉酮类化合物为式4所示化合物时,所述式4所示化合物由式3所示化合物制备得到,式3所示化合物制备式4所示化合物的反应式如下所示:
[0026][0027]
在本发明的第六方面,本发明提供一种在本发明第二方面所述的吡咯啉酮类化合物中间体的制备方法,当r3为乙烯基,r4为羟基取代甲基时,即所述吡咯啉酮类化合物为式3所示化合物时,所述式3所示化合物由式2所示化合物制备得到,式2所示化合物制备式3所示化合物的反应式如下所示:
[0028][0029]
在本发明的一个或多个实施例中,所述式2所示化合物由式5所示化合物制备得到,式5所示化合物制备式2所示化合物的反应式如下所示:
[0030][0031]
与现有技术相比,本发明具有以下优点和有益效果:
[0032]
1、本发明提供一种吡咯啉酮类化合物及其中间体,其可以作为药物中间体使用,特别是可以作为胆色素(包括胆红素)的中间体(a环)使用。
[0033]
2、本发明提供一种吡咯啉酮类化合物及其中间体的合成方法,特别是提供了3-甲基-4-乙烯基吡咯啉酮的合成方法,其合成路线短,适合工业化,成本低,合成条件温和。
[0034]
3、本发明提供的吡咯啉酮类化合物及其中间体的合成方法避免使用氰化钠等剧毒物质,环境友好。
具体实施方式
[0035]
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。使用的方法如无特别说明,均为本领域公知的常规方法,使用的耗材和试剂如无特别说明,均为市场购得。除非另有说明,本文中所用的专业与科学术语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法或材料也可应用于本发明中。nmr用bruker-amx600核磁共振仪测定;esi-ms用finnigan-mat-95质谱仪测定;所有试剂都为分析纯。
[0036]
本发明提供吡咯啉酮类化合物、吡咯啉酮类化合物中间体及其制备方法
[0037]
吡咯啉酮类化合物
[0038]
本发明提供一种吡咯啉酮类化合物,所述吡咯啉酮类化合物的结构式如式1所示,
[0039][0040]
其中,r1为c1~c12烷基;r2取自氢、苄基、取代苄基中的一种;优选地,r1为乙烯基或α对甲基苯磺酰基乙基,r2为氢或对甲氧基苄基或苄基。
[0041]
吡咯啉酮类化合物中间体
[0042]
在本发明的第二方面,本发明提供一种吡咯啉酮类化合物中间体,所述吡咯啉酮类化合物中间体的结构式如式6所示,
[0043][0044]
其中,r2取自氢、苄基、取代苄基中的一种,r3取自乙烯基或ch3ch,r4取自羟基取代甲基或醛基;优选地,r2为对甲氧基苄基或苄基。
[0045]
吡咯啉酮类化合物的制备方法
[0046]
在本发明的第三方面,本发明提供一种吡咯啉酮类化合物的制备方法,所述的吡咯啉酮类化合物如式1a所示,式1a所示化合物由式1b所示化合物制备得到,式1b所示化合物制备式1a所示化合物的反应式如下所示:
[0047][0048]
其中,r2取自苄基、取代苄基中的一种;优选地,r2为对甲氧基苄基或苄基。
[0049]
优选地,式1b所示化合物与对甲苯磺酸反应,纯化,得到所述式1a所示化合物,优
选地,控制反应温度为100~120摄氏度。
[0050]
在本发明的第四方面,本发明提供一种在本发明第一方面所述的另一种吡咯啉酮类化合物的制备方法,其特征在于,式1所示化合物由式4所示化合物制备得到,式4所示化合物制备式1所示化合物的反应式如下所示:
[0051][0052]
其中,r2取自苄基、取代苄基中的一种,r1为c1~c12烷基;优选地,r2为对甲氧基苄基或苄基,r1为乙烯基或α对甲基苯磺酰基乙基。
[0053]
优选地,式4所示化合物制备式1所示化合物的反应式如下所示:
[0054]
其中,r2为对甲氧基苄基或苄基。
[0055]
具体地,式4所示化合物与乙酸反应,纯化,得到所示式1b所示化合物,优选地,控制反应温度为100~120摄氏度。
[0056]
在另一些实施例中,式4所示化合物制备式1所示化合物的反应式如下所示:
[0057]
其中,r2为对甲氧基苄基或苄基。
[0058]
具体地,式4所示化合物、对甲苯亚磺酸钠、乙酸反应,纯化,得到所示式1c所示化合物,优选地,控制反应温度为50~70摄氏度。
[0059]
在本发明的一个或多个实施例中,所述式4所示化合物由式3所示化合物制备得到,式3所示化合物制备式4所示化合物的反应式如下所示:
[0060][0061]
具体地,式3所示化合物,三乙胺反应,得到式4所示化合物,优选地,控制反应温度为15~35℃,优选地,反应所使用的溶剂为dmso和二氯甲烷的混合溶剂。
[0062]
在本发明的一个或多个实施例中,所述式3所示化合物由式2所示化合物制备得到,式2所示化合物制备式3所示化合物的反应式如下所示:
[0063][0064]
具体地,式2所示化合物和4-甲氧基苄氨或苄胺反应,纯化,得到式3所示化合物。
[0065]
在本发明的一个或多个实施例中,所述式2所示化合物由式5所示化合物制备得到,式5所示化合物制备式2所示化合物的反应式如下所示:
[0066][0067]
具体地,式5所示化合物和原丙酸三乙酯反应,纯化,得到式2所示化合物。优选地,控制反应温度为90~160摄氏度。
[0068]
吡咯啉酮类化合物中间体的制备方法
[0069]
在本发明的第五方面,本发明提供一种在本发明第二方面所述的吡咯啉酮类化合物中间体的制备方法,当r3为ch3ch,r4为醛基时,即所述吡咯啉酮类化合物为式4所示化合物时,所述式4所示化合物由式3所示化合物制备得到,式3所示化合物制备式4所示化合物的反应式如下所示:
[0070][0071]
在本发明的第六方面,本发明提供一种在本发明第二方面所述的吡咯啉酮类化合物中间体的制备方法,当r3为乙烯基,r4为羟基取代甲基时,即所述吡咯啉酮类化合物为式3所示化合物时,所述式3所示化合物由式2所示化合物制备得到,式2所示化合物制备式3所示化合物的反应式如下所示:
[0072][0073]
在本发明的一个或多个实施例中,所述式2所示化合物由式5所示化合物制备得到,式5所示化合物制备式2所示化合物的反应式如下所示:
[0074][0075]
实施例1:化合物2的合成
[0076]
反应式如下所示:
[0077][0078]
具体制备过程如下:2000毫升三口瓶中,投入409克(4.64mol)顺丁烯二醇(式5所示化合物)和900克(5.11mol)原丙酸三乙酯,加热搅拌9小时,温度由100度升到155度,乙醇不断蒸馏出,直到没有液体蒸馏出,降温到75度,连接一个15cm高的玻璃珠填料精馏柱,收集bp 75℃(11mmhg)的馏份,得到552克产物(无色液体),即为式2所示化合物,收率94%。
[0079]
实施例2:化合物n-【(4-甲氧基苯基甲基)】-2-甲基-3-羟甲基-4-戊烯酰胺(式3所示化合物)的合成
[0080]
反应式如下所示:
[0081][0082]
其中,r2为对甲氧基苄基。
[0083]
具体制备过程如下:50.0克(0.397mol)式2所示化合物和4-甲氧基苄氨(217.3克,1.58mol)在氮气保护下加热搅拌20小时,过量的苄胺减压蒸馏回收(油浴温度95度,水泵抽真空),加入127毫升乙酸乙酯和90毫升石油醚,15~35℃下搅拌一小时,析出大量固体,过滤,用1:1ea:pe洗涤,五氧化二磷真空干燥,得到白色固体71.8克,即为式3所示化合物,收率68.7%,mp:80~82℃,1h nmr(cdcl3)δ7.22(d,2h),6.88(d,2h),5.99(s,1h),5.74(m,1h),5.20(m,2h),4.39(m,2h),3.82(s,3h),3.64(m,2h),2.45(m,1h),2.37(m,1h),1.19(d,1h).ms m/z 263(m
).
[0084]
实施例3:化合物n-苄基-2-甲基-3-羟甲基-4-戊烯酰胺的合成
[0085]
反应式如下所示:
[0086][0087]
其中,r2为苄基。
[0088]
具体制备过程如下:21.6克(0.171mol)式2所示化合物和苄氨(36.7克,0.342mol,2eqv)在氮气保护下加热搅拌20小时,过量的苄胺减压蒸馏回收(油浴温度95度,水泵抽真空),加入乙酸乙酯和石油醚重结晶后得到白色固体17.7克,即为式3所示化合物,收率44.3%。
[0089]
实施例4:化合物n-【(4-甲氧基苯基甲基)】-2-甲基-3-氧代甲基-4-戊烯酰胺(式4所示化合物)的合成
[0090]
反应式如下所示:
[0091][0092]
其中,r2为对甲氧基苄基。
[0093]
具体制备过程如下:式3所示化合物26.3克(100mmol)、40.4克三乙胺(400mmol)、dmso(156.3克,2000mmol)的二氯甲烷溶液(260毫升),在15~35℃搅拌下用时15分钟加入三氧化硫吡啶复合物(48.0克,301mmol),得到棕色反应液,15~35℃继续搅拌一小时后加入900毫升4n盐酸淬灭反应,用dcm萃取,水洗涤,然后碳酸氢钠洗涤,盐水洗涤,硫酸钠干燥,旋蒸除溶剂后得到24.1克浅黄色油,即为式4所示化合物,收率92.3%。1h nmr(cdcl3)δ9.34(s,1h),7.17(d,2h),6.86(d,2h),6.72(q,1h),6.07(s,1h),4.35(m,2h),3.80(s,3h),3.72(q,1h),2.09(d,3h),1.38(d,3h).
[0094]
实施例5:化合物n-苄基-2-甲基-3-氧代甲基-4-戊烯酰胺(式4所示化合物)的合成
[0095]
反应式如下所示:
[0096][0097]
其中,r2为苄基。
[0098]
具体制备过程如下:式3所示化合物2.00克(8.58mmol)、3.46克三乙胺(34.3mmol)、dmso(13.38克,171.6mmol)的二氯甲烷溶液(20毫升),在15~35℃搅拌下用时15分钟加入三氧化硫吡啶复合物(4.08克,25.7mmol),得到棕色反应液,15~35℃继续搅拌一小时后加入80毫升4n盐酸淬灭反应,用dcm萃取,水洗涤,然后碳酸氢钠洗涤,盐水洗涤,硫酸钠干燥,旋蒸除溶剂后得到1.9克浅黄色油,即为式4所示化合物,收率95.8%。
[0099]
实施例6:化合物n-【(4-甲氧基苯基甲基)】-3-甲基-4-乙烯基-1,5二氢-2h-吡咯啉-2-酮(式1b所示化合物)的合成
[0100]
反应式如下所示:
[0101][0102]
其中,r2为对甲氧基苄基。
[0103]
具体制备过程如下:4.1克式4所示化合物(15.7mmol)和40毫升乙酸在氮气保护下110度加热搅拌20小时,旋蒸除乙酸,ea溶解残留物,水洗,然后碳酸氢钠洗涤,盐水洗涤,硫酸钠干燥,旋蒸除溶剂后得浅黄色油,用ea:pe 2:1重结晶得到2.6克浅黄固体,即为式1b所
示化合物,mp:58~60℃,收率68%。1h nmr(cdcl3)δ1.80(s,3h),3.76(s,3h),3.84(s,2h),4.58(s,2h),5.27~5.33(m,2h),6.66~6.71(m,1h),6.85(m,2h),7.19(m,2h)。
[0104]
实施例7:化合物n-苄基-3-甲基-4-乙烯基-1,5二氢-2h-吡咯啉-2-酮(式1b所示化合物)的合成
[0105]
反应式如下所示:
[0106][0107]
其中,r2为苄基。
[0108]
具体制备过程如下:1.0克式4所示化合物(4.3mmol)和10毫升乙酸在氮气保护下110度加热搅拌2小时,旋蒸除乙酸,ea溶解残留物,水洗,然后碳酸氢钠洗涤,盐水洗涤,硫酸钠干燥,旋蒸除溶剂后得浅黄色油,用ea:pe 4:1柱层析得到0.33克油状物,即为式1b所示化合物,收率35.8%。1h nmr(cdcl3)δ1.90(s,3h),3.87(m,2h),4.65(s,2h),5.30(m,2h),6.69(m,1h),7.26(m,5h).ms m/z 214(m
1).
[0109]
实施例8:化合物3-甲基-4-乙烯基
‑‑
1,5二氢-2h-吡咯啉-2-酮(式1a所示化合物)的合成
[0110]
反应式如下所示:
[0111][0112]
其中,r2为对甲氧基苄基。
[0113]
具体制备过程如下:式1b所示化合物(2.00克,8.2mmol)、6.25克(32.9mmol)对甲苯磺酸一水物和40毫升甲苯,在氮气保护下110度加热搅拌5小时,冷却到15~35℃,水洗,然后碳酸氢钠洗涤,盐水洗涤,硫酸钠干燥,旋蒸除溶剂后得浅黄色油,用ea:pe 1:3重结晶得到0.66克浅黄固体,即为式1a所示化合物,mp:85~87℃(文献86~88度),收率65%,ms m/z 123(m
).核磁与文献一致。
[0114]
实施例9:化合物n-【(4-甲氧基苯基甲基)】-3-甲基-4-(1-对甲基苯磺酰基乙基)-1,5二氢-2h-吡咯啉-2-酮(1c)的合成
[0115][0116]
其中,r2为对甲氧基苄基。
[0117]
具体制备过程如下:8.53克(32.6mmol)式4所示化合物、5.81克(32.6mmol)对甲苯
亚磺酸钠、2.94克乙酸(49.0mmol)、240毫升乙醇、40毫升水的混合物在氮气保护下于60度加热搅拌4小时,旋蒸除溶剂,残留物用乙酸乙酯溶解,水洗,然后碳酸氢钠洗涤,盐水洗涤,硫酸钠干燥,旋蒸除溶剂后得黄色膏状物,用1:2ea:pe重结晶得类白色固体5.45克,即为式1c所示化合物,收率43%。1h nmr(cdcl3)δ7.57(d,2h),7.29(d,2h),7.16(d,2h),6.87(d,2h),4.59(d,j=14.80hz,1h),4.52(d,j=14.80hz,1h),4.21(q,j=7.2hz,1h),3.99(d,j=19.2hz,1h),3.81(s,3h),3.67(d,j=19.2hz,1h),2.43(s,3h),1.57(d,j=7.2hz,3h),1.34(s,3h).ms m/z 399(m
).
[0118]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。