1.本发明属于植物有效成分提取技术领域,具体涉及一种新克罗地烷型二萜化合物及其制备方法和应用。
背景技术:
2.巴豆属(croton)植物中含有丰富的活性成分。我国目前发现该属植物有20多种,主要分布在我国东部和西南地区。该属植物中有很多种都可入药,较为常见的药用部位是根、枝叶和果(种仁)。其中根茎具有较强的杀虫、杀菌生物活性,枝叶具有较好的抗肿瘤、抗炎镇痛作用,种仁具有促泄、刺激平滑肌和加强胃排空等功效。我们在前期降糖活性筛选过程中,发现云南巴豆的90%乙醇提取物具有显著的降糖活性。
3.糖尿病是目前严重危害人类健康的三大疾病之一,当前临床一线虽然较多,且靶点多样,但是这些降糖药物主要为西药,同时存在毒副作用大,长期服药对患者造成严重的经济负担。
4.为此,本发明从巴豆属植物云南巴豆中分离得到一个结构新颖的新克罗地烷型二萜crotonyunnansine f,该化合物具有能显著抑制钠-葡萄糖共转运蛋白2(sglt-2)的活性,并能提高glut-4的表达。
技术实现要素:
5.为了解决上述技术问题,本发明提供了一种新克罗地烷型二萜化合物及其制备方法和应用,于巴豆中获得新结构的克罗地烷型二萜crotonyunnansine f,应用于糖尿病治疗,具有抑制钠-葡萄糖共转运蛋白2(sglt-2)的活性,并能提高glut-4的表达。
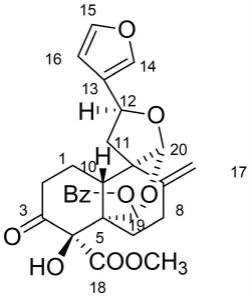
6.为了达到解决上述技术问题的技术效果,本发明是通过以下技术方实现的:一种新克罗地烷型二萜化合物,其特征在于,化学结构如式为:
[0007][0008]
本发明的又一目的在于提供一种新克罗地烷型二萜化合物的制备方法,其特征在于,以干燥的巴豆属植物枝叶及果(种仁)为原料,经浸膏提取、有机溶剂萃取、硅胶柱层析、高压液相色谱分离获得,具体包括以下步骤:
[0009]
s1、浸膏提取:将云南巴豆枝叶及果(种仁)粉碎,用有机溶剂回流提取,并获得提取液;而后将提取液过滤,并进行减压浓缩操作,静置滤除沉淀物后,浓缩成浸膏a;
[0010]
s2、有机溶剂萃取:在上述s1中的到浸膏a中加入重量比1~2倍量的水,用与水等体积的有机溶剂萃取,而后合并有机溶剂萃取相,减压浓缩成浸膏b;
[0011]
s3、硅胶柱层析:将浸膏b用重量比1.5~3倍量的氯仿溶解,用浸膏重0.8~1.2倍的80~100目硅胶拌样,然后上硅胶柱层析;用体积比为1:0~0:1的混合有机溶剂对硅胶柱层析的物质进行梯度洗脱,收集梯度洗脱液并浓缩,经tlc检测,合并相同的部分;
[0012]
s4、反相柱层析:将s3中洗脱得到的洗脱液上反相柱层析;用体积含量为20~100%的甲醇水溶液对反相柱层析的物质进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc检测,合并相同的部分;
[0013]
s5、高效液相色谱分离:将s4中以体积含量在70~85%内的甲醇水溶液洗脱得到的洗脱液经高效液相色谱分离纯化,得到新克罗地烷型二萜类化合物crotonyunnansine f;
[0014]
进一步的,所述s1中将云南巴豆枝叶及果(种仁)粉碎到20~40目,且采用有机溶剂在85-90℃下回流提取2~3次;
[0015]
进一步的,所述s1中减压浓缩将提取液浓缩至1/4~1/2体积;
[0016]
进一步的,所述s2中有机溶剂萃取3~4次;
[0017]
进一步的,所述s3中装柱硅胶为160~200目,用量为浸膏b重量6~8倍量;
[0018]
进一步的,所述s4中反相柱是用反相材料c-18装柱
[0019]
进一步的,所述s5中的高效液相色谱分离纯化以75%的甲醇为流动相,用流速2ml/min,10
×
250mm(5μm)的agilent zorbax c18制备色谱柱为固定相,紫外检测器检测波长为254nm;
[0020]
进一步的,所述高效液相色谱分离中每次进样50~100μl,收集17.5~20.0min的色谱峰,多次累加后蒸干,即得所述的新克罗地烷型二萜类化合物crotonyunnansine f;
[0021]
本发明的有益效果是:
[0022]
本发明从巴豆属植物云南巴豆中分离得到一新的新克罗地烷型二萜crotonyunnansine f,该化合物具有显著抑制钠-葡萄糖共转运蛋白2(sglt-2)的活性,并能提高glut-4的表达,
附图说明
[0023]
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0024]
图1是一种新克罗地烷型二萜化合物crotonyunnansine f的化学结构示意图;
[0025]
图2为本发明化合物crotonyunnansine f的核磁共振氢谱(1hnmr);
[0026]
图3为本发明化合物crotonyunnansine f的核磁共振碳谱(13cnmr);
[0027]
图4为本发明化合物crotonyunnansine f的单晶衍射结构图(铜靶);
[0028]
图5是本发明化合物crotonyunnansine f制备的结果分析图。
具体实施方式
[0029]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0030]
实施例1
[0031]
一种新克罗地烷型二萜化合物,其特征在于,化学结构如式为:
[0032][0033]
实施例2
[0034]
一种新克罗地烷型二萜化合物的制备方法,其特征在于,以干燥的巴豆属植物枝叶及果(种仁)为原料,经浸膏提取、有机溶剂萃取、硅胶柱层析、高压液相色谱分离获得,具体包括以下步骤:
[0035]
步骤1、浸膏提取:将云南巴豆枝叶及果(种仁)粉碎到20~40目,用有机溶剂在85-90℃下回流提取2~3次,并获得提取液;而后将提取液过滤,并进行减压浓缩操作,静置滤除沉淀物后,浓缩成浸膏a,其中减压浓缩将提取液浓缩至1/4~1/2体积;
[0036]
步骤2、有机溶剂萃取:在上述s1中的到浸膏a中加入重量比1~2倍量的水,用与水等体积的有机溶剂萃取3到4次,而后合并有机溶剂萃取相,减压浓缩成浸膏b;
[0037]
步骤3、硅胶柱层析:将浸膏b用重量比1.5~3倍量的氯仿溶解,用浸膏重0.8~1.2倍的80~100目硅胶拌样,然后上硅胶柱层析,装柱硅胶为160~200目,用量为浸膏b重量6~8倍量;用体积比为1:0~0:1的混合有机溶剂梯度洗脱,收集梯度洗脱液并浓缩,经tlc检测,合并相同的部分;
[0038]
步骤4、反相柱层析:将s3中洗脱得到的洗脱液上反相柱层析;用体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc检测,合并相同的部分;
[0039]
步骤5、高效液相色谱分离:将s4中得到的洗脱液(以体积含量70~85%甲醇水溶液洗脱得到的洗脱液)经高效液相色谱分离纯化,得到新克罗地烷型二萜类化合物crotonyunnansine f;其中中的高效液相色谱分离纯化以75%的甲醇为流动相,用流速2ml/min,10
×
250mm(5μm)的agilent zorbax c18制备色谱柱为固定相,紫外检测器检测波长为254nm;在高效液相色谱分离中每次进样50~100μl,收集17.5~20.0min的色谱峰,多次累加后蒸干,即得所述的新克罗地烷型二萜类化合物crotonyunnansine f。
[0040]
实施例3
[0041]
本实施例为基于上述实施例一种新克罗地烷型二萜化合物的制备方法的新克罗
地烷型二萜类化合物crotonyunnansine f的制备实验;
[0042]
实验1
[0043]
取干燥的巴豆属植物云南巴豆(c.yunnanensis)植物枝叶及果(种仁)10.0kg,粗粉碎至40目,用90%的乙醇在85-90℃下回流提取3次,每次60min,提取液合并;提取液过滤,减压浓缩至体积的1/4;静置,滤除沉淀物,浓缩成约1123g浸膏a;在浸膏a中加入1123g水,用与水等体积的石油醚萃取3次,合并萃取相,减压浓缩成400g浸膏b;用200目硅胶2800g装柱,在浸膏b中加入800g的氯仿溶解,然后加入90目硅胶480g拌样,室温挥干后上柱;用体积比分别为1:0、20:1、10:1、8:2、3:2、1:1、1:2、0:1的石油醚-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分,得到8个部分,体积比8:2的石油醚-丙酮混合有机溶剂的洗脱液c为31g;用反相材料c-18装柱,洗脱液c(31g)上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量70~85%甲醇水溶液洗脱得到的洗脱液,再以75%的甲醇为流动相,流速2ml/min,10
×
250mm(5μm)的agilentzorbax c18制备色谱柱为固定相,紫外检测器检测波长为254nm,每次进样50μl,收集20.0min的色谱峰,多次累加后蒸干,即得所述的新克罗地烷型二萜类化合物crotonyunnansine f。
[0044]
实验2
[0045]
取干燥的巴豆属植物曼哥龙巴豆(c.mangelong)植物枝叶及果(种仁)8kg,粗粉碎至20目,用80%的丙酮在85-90℃下回流提取2次,每次50min,提取液合并;提取液过滤,减压浓缩至体积的1/3;静置,滤除沉淀物,浓缩成800g浸膏a;在浸膏a中加入800g的水,用与水等体积的氯仿萃取3次,合并萃取相,减压浓缩成309g浸膏b;用160目硅胶2472g装柱,在浸膏b中加入927g的氯仿溶解,然后加入80目硅胶247.2g拌样,室温挥干后上柱;用体积比分别为1:0、20:1、10:1、8:2、3:2、1:1、1:2、0:1的正己烷-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;体积比10:1的石油醚-丙酮混合有机溶剂的洗脱液c为25g;用反相材料c-18装柱,洗脱液c上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量70~85%甲醇水溶液洗脱得到的洗脱液,再以75%的甲醇为流动相,流速2ml/min,10
×
250mm(5μm)的agilent zorbax c18制备色谱柱为固定相,紫外检测器检测波长为254nm,每次进样70μl,收集20min的色谱峰,多次累加后蒸干得新克罗地烷型二萜类化合物crotonyunnansine f。
[0046]
实验3
[0047]
取干燥的巴豆属植物曼哥龙巴豆(c.mangelong)枝叶及果(种仁)6kg,粗粉碎至30目,用90%的甲醇在85-90℃下回流提取3次,每次30min,提取液合并;提取液过滤,减压浓缩至体积的1/2;静置,滤除沉淀物,浓缩成680g浸膏a;在浸膏a中加入1360g的水,用与水等体积的乙醚萃取4次,合并萃取相,减压浓缩成210g浸膏b;用200目硅胶1680g装柱,在浸膏b中加入420g的氯仿溶解,然后加入100目硅胶210g拌样,室温挥干后上柱;用体积比分别为1:0、20:1、10:1、8:2、3:2、1:1、1:2、0:1的氯仿-丙酮混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;体积比10:1的氯仿-丙酮混合有机溶剂的洗脱液c为17g。用反相材料c-18装柱,洗脱液c上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量70~85%
甲醇水溶液洗脱得到的洗脱液,再以75%的甲醇为流动相,流速3ml/min,10
×
250mm(5μm)的agilent zorbax c18制备色谱柱为固定相,紫外检测器检测波长为254nm,每次进样80μl,收集17.5min的色谱峰,多次累加后蒸干即得新克罗地烷型二萜类化合物crotonyunnansine f。
[0048]
实验4
[0049]
取干燥的巴豆属植物云南巴豆(c.yunnanensis)枝叶及果(种仁)5kg,粗粉碎至20目,用100%甲醇在85-90℃下回流提取4次,每次35min,提取液合并;提取液过滤,减压浓缩至体积的1/2;静置,滤除沉淀物,浓缩成508g浸膏a;在浸膏a中加入1016g的水,用与水等体积的苯萃取5次,合并萃取相,减压浓缩成196g浸膏b;用200目硅胶1372g装柱,在浸膏b中加入588g的氯仿溶解,然后加入100目硅胶156.8g拌样,室温挥干后上柱;用体积比分别为1:0、20:1、10:1、8:2、3:2、1:1、1:2、0:1的石油醚-乙酸乙酯混合有机溶剂梯度洗脱,收集梯度洗脱液、浓缩,经tlc监测,合并相同的部分;体积比10:1的石油醚-乙酸乙酯混合有机溶剂的洗脱液c为15.2g。用反相材料c-18装柱,洗脱液c上反相柱,以体积含量为20~100%的甲醇水溶液进行梯度洗脱,收集各部分洗脱液并浓缩,经tlc监测,合并相同的部分;取以体积含量70~85%甲醇水溶液洗脱得到的洗脱液,再以75%的甲醇为流动相,流速3ml/min,10
×
250mm(5μm)的agilent zorbax c18制备色谱柱为固定相,紫外检测器检测波长为254nm,每次进样100μl,收集17.5min的色谱峰,多次累加后蒸干,得新克罗地烷型二萜类化合物crotonyunnansine f。
[0050]
实施例4
[0051]
本实施例为上述实施例3中各实验制备的新克罗地烷型二萜类化合物crotonyunnansine f的鉴定;
[0052]
取实验1制备的化合物crotonyunnansine f,为白色无定型粉末;测定方法为:用核磁共振,结合其它波谱技术鉴定结构。
[0053]
(1)esi-ms( ):m/z 531[m na] .hresims( )显示本发明化合物准分子离子峰m/z 531.1628[m na] (c28h28o9na ,计算值为531.1631)。
[0054]
(2)1h nmr(cdcl3,400mhz)和13c nmr(cdcl3,100mhz)数据,见表1。
[0055]
表1化合物crotonyunnansine f的1h(400mhz)和13c nmr(100mhz)数据(溶剂为cdcl3),化学位移δ(ppm),偶合常数j(hz)。
[0056]
表1
[0057][0058][0059]
取实验2制备的化合物crotonyunnansine f分别按上述方法进行结构测定,结果为:其结构同实施例5,分子式为c28h28o9。
[0060]
取实验3制备的化合物crotonyunnansine f分别按上述方法进行结构测定,结果为:其结构同实施例5,分子式为c28h28o9。
[0061]
取实验4制备的化合物biperovskatone分别按上述方法进行结构测定,结果为:其结构同实施例5,分子式为c28h28o9。
[0062]
取实验1~4所制备的任一的新克罗地烷型二萜类化合物crotonyunnansine f进行降糖活性检测试验,试验情况如下:
[0063]
(1)cho细胞sglt-2抑制活性
[0064]
实验细胞株:
[0065]
采用标准技术克隆人类sglt-2全长互补脱氧核糖核酸(cdna)序列并稳定转染到中国仓鼠卵巢细胞(cho)中。采用放射性标记的葡萄糖类似物[14c]-甲基-α-d-吡喃葡萄糖苷([14c]-amg,perkin elmer)作为转运体底物。
[0066]
缓冲液:
[0067]
配制实验缓冲液,模拟肾小球滤过的低蛋白的条件。缓冲液包括:10mm羟乙基哌嗪乙硫磺酸(hepes),1.2mm氯化镁(mgcl2),120mm氯化钠(nacl),4.7mm氯化钾(kcl)和2.2mm氯化钙(cacl2),ph=7.4。并使用该缓冲液配制6μm的[14c]-amg缓冲液。
[0068]
sglt-2体外抑制活性测试:
[0069]
将能够稳定表达人sglt-2基因的cho细胞接种到含f12(1x)培养基(invitrogen)的96孔细胞培养板中,在37℃,5%co2培养箱中过夜。使用ph=7.4的实验缓冲液冲洗细胞后,用49μl缓冲液、1μl的待测化合物的dmso稀释液和50μl的6μm的[14c]-amg缓冲液。在37℃条件下孵育细胞1h。
[0070]
通过去除培养液,停止摄取反应,用冰水终止缓冲液冲洗细胞停止孵化。然后加入50μl 10%naoh冰冷裂解缓冲液裂解细胞,将细胞裂解物转入picoproas-vial,加入2ml的ultima gold cocktail。用tri-carb对细胞内的放射性含量进行量化,并使用graphppad prism 5.0软件对所得到的数据分析。调整响应曲线拟合经验四参数模型,确定待测化合物的半数最大响应浓度,即ic50。阳性对照达格列净的剂量响应曲线同时测定。
[0071]
(2)对胰岛素抵抗的3t3-l1细胞中glut-4蛋白的表达影响检测
[0072]
实验细胞株:3t3-l1前脂肪细胞,采用地塞米松(1μm)和胰岛素(10nm)联合诱导方式方式获得胰岛素抵抗的3t3-l1细胞模型。
[0073]
胰岛素抵抗模型:为了验证获得胰岛素抵抗细胞,对以上细胞设定四个实验组,第1、2、3、4组分化的3t3-l1细胞进行处理:除第1组阴性对照加正常培养基以外,第2组加入1μm的地塞米松,第3、4组同时分别加入1μm的地塞米松和10nm的胰岛素,作用4天,每2天换液一次,取培养上清液,用临床糖检测试剂葡萄糖氧化酶-过氧化物酶法(god-pod)法(该方法为微量化测定法)检测培养上清液中的葡萄糖含量。
[0074]
(3)western blot法检测ir-3t3-l1脂肪细胞glut4蛋白表达水平:
[0075]
pbs将细胞洗3遍,加入裂解液,置冰上15min,充分裂解后,4℃14000r/min离心15min,取上清液即为总蛋白。用bca测定蛋白浓度,根据蛋白浓度取各组样本(20μg),以10%十二烷基硫酸钠聚丙烯酰胺凝胶(sds-page)分离蛋白,然后以湿转法(280ma,1h)将蛋白质转移至pvdf膜上,再用含5%脱脂奶粉的1
×
tbst室温下封闭1h后,加入一抗,4℃过夜后,1
×
tbst洗膜3次,每次10min;加入辣根过氧化物酶标记的二抗,室温轻摇1h;充分洗涤后加入ecl工作液,放入化学发光凝胶成像仪中设置程序显影;采用bio-rad公司的image lab软件分析。
[0076]
(3)实验结果
[0077]
实验结果表明:经检测其对钠-葡萄糖共转运蛋白2(sglt-2)抑制活性的ic50值达1.98nm;且在0.12μm时能显著提高ir-3t3-l1细胞中glut-4的表达,提示该化合物具有显著的降糖活性,如要图5所示;
[0078]
图5为化合物crotonyunnansine f在0.24μm时对ir-3t3-l1细胞中glut-4蛋白表达的影响。1-正常组;2-空白组;3-阳性对照(罗格列酮)组;4-crotonyunnansine f组。
[0079]
综上所述,本发明从巴豆属植物云南巴豆中分离得到一新的新克罗地烷型二萜crotonyunnansine f,该化合物具有显著抑制钠-葡萄糖共转运蛋白2(sglt-2)的活性,并能提高glut-4的表达
[0080]
在本说明书的描述中,参考术语“一个实施例”、“示例”、“具体示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
[0081]
以上公开的本发明优选实施例只是用于帮助阐述本发明。优选实施例并没有详尽叙述所有的细节,也不限制该发明仅为所述的具体实施方式。显然,根据本说明书的内容,可作很多的修改和变化。本说明书选取并具体描述这些实施例,是为了更好地解释本发明的原理和实际应用,从而使所属技术领域技术人员能很好地理解和利用本发明。本发明仅受权利要求书及其全部范围和等效物的限制。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。