1.本发明涉及高分子材料技术领域,尤其涉及一种聚酯组合物、其制备方法及应用。
背景技术:
2.当前,由于环境保护和能源紧缺,生物基可生物降解聚酯获得了大力的发展。聚乳酸(pla)是一种生物基可生物降解聚酯,具有良好的拉伸强度,但断裂伸长率低,限制了其应用。同时,聚乳酸的阻隔性能很差,不能应用到阻隔要求高的领域,例如,食品包装和药品包装领域。同时,介电常数低。
3.因此,开发韧性、阻隔性能高的全生物基、可生物降解聚酯具有重要意义。
技术实现要素:
4.有鉴于此,本发明要解决的技术问题在于提供一种聚酯组合物、其制备方法及应用,本发明提供的聚酯组合物可生物降解,力学性能、阻隔性能较优,介电性能较优。
5.本发明提供了一种聚酯组合物,按重量份数计包括:
6.聚乳酸
ꢀꢀꢀꢀꢀꢀꢀ
5~95份;
7.聚酯s
ꢀꢀꢀꢀꢀꢀꢀꢀ
5~95份;
8.所述聚酯s具有式ⅰ所示结构:
[0009][0010]
式ⅰ中,m≥1,n≥1;
[0011]
r1具有式ⅱ所示结构;
[0012]-(ch2)
p
‑ꢀꢀꢀ
式ⅱ;
[0013]
式ⅱ中,1≤p≤8;
[0014]
r2具有式iii所示结构;
[0015][0016]
式iii中,r3选自硫或氧。
[0017]
优选的,1000≥m≥1,1000≥n≥1。
[0018]
优选的,式ⅱ中,p为2、3、4、7或8。
[0019]
优选的,r2选自式iii-1~式iii-3所示结构中的至少一种;
[0020][0021]
优选的,所述聚酯s按照以下方法进行制备:
[0022]
a)在保护气的条件下,将物料a、物料b和物料c在催化剂的作用下进行反应,得到预聚物;
[0023]
所述物料a包括2,5-呋喃二甲酸或其衍生物、2,4-呋喃二甲酸或其衍生物和2,5-噻吩二甲酸或其衍生物中的一种;
[0024]
所述物料b包括1,4-丁二酸、1,5-戊二酸、1,6-己二酸、1,9-壬二酸或1,10-癸二酸;
[0025]
所述物料c包括1,4-丁二醇;
[0026]
b)在真空条件下,将所述预聚物进行缩聚反应,得到聚酯s。
[0027]
优选的,步骤a)中,所述物料a包括2,4-呋喃二甲酸、2,4-呋喃二甲酸二甲酯、2,4-呋喃二甲酸二乙酯、2,5-呋喃二甲酸、2,5-呋喃二甲酸二甲酯、2,5-呋喃二甲酸二乙酯、2,5-噻吩二甲酸、2,5-噻吩二甲酸二甲酯或2,5-噻吩二甲酸二乙酯;
[0028]
所述催化剂包括氧化亚锡、辛酸亚锡、氯化亚锡、溴化亚锡、碘化亚锡、乙酸亚锡、草酸亚锡、硫酸亚锡、氢氧化亚锡、钛酸四丁酯、钛酸异丁酯、钛酸正丙酯、钛酸异丙酯、氧化锌、乙酸锌、锌、乙酰丙酮铝、醋酸锑、乙二醇锑、三氧化二锑和二氧化锗中的一种或两种。
[0029]
优选的,步骤a)中,物料a和物料b的摩尔比为0.01~7:0.01~4;
[0030]
所述物料c占所述物料a和物料b的摩尔总和的摩尔比为0.01~3:0.01~1;
[0031]
所述催化剂占所述物料a和物料b的摩尔总和的摩尔比为0.01%~10%。
[0032]
优选的,步骤a)中,所述反应的温度为150~300℃,时间为0.5~5h,压力为0.01~0.5mpa;
[0033]
步骤b)中,所述缩聚反应的温度为150~300℃,时间为0.5~10h,真空度为5~100pa。
[0034]
本发明还提供了一种上文所述的聚酯组合物的制备方法,包括以下步骤:
[0035]
将聚乳酸和聚酯s混合后,经熔融挤出造粒,得到聚酯组合物。
[0036]
本发明还提供了一种聚酯组合物作为阻隔包装材料或介电材料的应用;
[0037]
所述聚酯组合物为上文所述的聚酯组合物,或为上文所述的制备方法制得的聚酯组合物。
[0038]
本发明制备的全生物基聚酯组合物具有良好的韧性和阻隔性能。其断裂伸长率从纯pla的5%提高到了100%以上,无缺口冲击强度由纯pla的14kj/m2提高至35kj/m2以上;与纯pla相比,全生物基聚酯组合物的氧气透过率也大幅度降低,氧气透过系数低于150cm3/m2·
24h
·
0.1mpa,说明其阻隔性明显变好。与纯pla相比,全生物基聚酯组合物的拉伸强度也略有增加。同时,聚酯组合物的介电常数也相应增加。同时,本发明的原料组分都可从生物质转化制备,可生物降解,可摆脱其对石油资源的依赖。
附图说明
[0039]
图1为本发明实施例1的聚酯s的核磁氢谱图。
具体实施方式
[0040]
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,
本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0041]
本发明提供了一种聚酯组合物,按重量份数计包括:
[0042]
聚乳酸
ꢀꢀꢀꢀꢀꢀ
5~95份;
[0043]
聚酯s
ꢀꢀꢀꢀꢀꢀꢀ
5~95份;
[0044]
所述聚酯s具有式ⅰ所示结构:
[0045][0046]
式ⅰ中,m≥1,n≥1;
[0047]
r1具有式ⅱ所示结构;
[0048]-(ch2)
p
‑ꢀꢀꢀ
式ⅱ;
[0049]
式ⅱ中,1≤p≤8;
[0050]
r2具有式iii所示结构;
[0051][0052]
式iii中,r3选自硫或氧。
[0053]
本发明提供的聚酯组合物包括聚乳酸。在本发明的某些实施例中,所述聚酯组合物中,聚乳酸的重量份数为75份、60份、68份或65份。
[0054]
在本发明的某些实施例中,所述聚乳酸包括右旋聚乳酸(pdla)、左旋聚乳酸(plla)和外消旋聚乳酸(pdlla)中的至少一种。
[0055]
在本发明的某些实施例中,所述聚乳酸的重均分子量为20000~1000000。在某些实施例中,所述聚乳酸的重均分子量为254300。
[0056]
本发明提供的聚酯组合物还包括聚酯s。在本发明的某些实施例中,所述聚酯组合物中,聚酯的重量份数为25份、40份、32份或35份。
[0057]
所述聚酯s具有式ⅰ所示结构:
[0058][0059]
式ⅰ中,m≥1,n≥1。
[0060]
在本发明的某些实施例中,1000≥m≥1,1000≥n≥1。在某些实施例中,m=42、118、56或39,n=98、118、84或91。
[0061]
r1具有式ⅱ所示结构;
[0062]-(ch2)
p
‑ꢀꢀꢀ
式ⅱ;
[0063]
式ⅱ中,1≤p≤8。
[0064]
在本发明的某些实施例中,p为2、3、4、7或8。
[0065]
r2具有式iii所示结构;
[0066][0067]
式iii中,r3选自硫或氧。
[0068]
在本发明的某些实施例中,r2选自式iii-1~式iii-3所示结构中的至少一种;
[0069][0070]
在本发明的某些实施例中,所述聚酯s按照以下方法进行制备:
[0071]
a)在保护气的条件下,将物料a、物料b和物料c在催化剂的作用下进行反应,得到预聚物;
[0072]
所述物料a包括2,5-呋喃二甲酸或其衍生物、2,4-呋喃二甲酸或其衍生物和2,5-噻吩二甲酸或其衍生物中的一种;
[0073]
所述物料b包括1,4-丁二酸、1,5-戊二酸、1,6-己二酸、1,9-壬二酸或1,10-癸二酸;
[0074]
所述物料c包括1,4-丁二醇;
[0075]
b)在真空条件下,将所述预聚物进行缩聚反应,得到聚酯s。
[0076]
步骤a)中:
[0077]
在本发明的某些实施例中,所述保护气为惰性气体,具体可以为氮气。
[0078]
在本发明的某些实施例中,所述物料a包括2,4-呋喃二甲酸、2,4-呋喃二甲酸二甲酯、2,4-呋喃二甲酸二乙酯、2,5-呋喃二甲酸、2,5-呋喃二甲酸二甲酯、2,5-呋喃二甲酸二乙酯、2,5-噻吩二甲酸、2,5-噻吩二甲酸二甲酯或2,5-噻吩二甲酸二乙酯。
[0079]
在本发明的某些实施例中,所述催化剂包括锡类催化剂、钛类催化剂、锌类催化剂、铝类催化剂和锗类催化剂中的一种或两种。
[0080]
在本发明的某些实施例中,所述催化剂包括氧化亚锡、辛酸亚锡、氯化亚锡、溴化亚锡、碘化亚锡、乙酸亚锡、草酸亚锡、硫酸亚锡、氢氧化亚锡、钛酸四丁酯、钛酸异丁酯、钛酸正丙酯、钛酸异丙酯、氧化锌、乙酸锌、锌、乙酰丙酮铝、醋酸锑、乙二醇锑、三氧化二锑和二氧化锗中的一种或两种。
[0081]
在本发明的某些实施例中,物料a和物料b的摩尔比为0.01~7:0.01~4。在某些实施例中,物料a和物料b的摩尔比为7:3、3:3或6:4。
[0082]
在本发明的某些实施例中,所述物料c占所述物料a和物料b的摩尔总和的摩尔比为0.01~3:0.01~1。在某些实施例中,所述物料c占所述物料a和物料b的摩尔总和的摩尔比为3:1。
[0083]
在本发明的某些实施例中,所述催化剂占所述物料a和物料b的摩尔总和的摩尔比为0.01%~10%。在某些实施例中,所述催化剂占所述物料a和物料b的摩尔总和的摩尔比为0.3%。
[0084]
在本发明的某些实施例中,所述反应为酯化反应或酯交换反应。所述反应的温度为150~300℃,时间为0.5~5h,压力为0.01~0.5mpa。在某些实施例中,所述反应的温度为180℃,时间为4h,压力为0.01mpa。所述反应在搅拌的条件下进行。
[0085]
步骤b)中:
[0086]
在本发明的某些实施例中,所述缩聚反应的温度为150~300℃,时间为0.5~10h,真空度为5~100pa。在某些实施例中,所述缩聚反应的温度为230℃,时间为10h,真空度为5pa。
[0087]
缩聚反应后,得到聚酯s。
[0088]
在本发明的某些实施例中,所述聚酯s的数均分子量为10000~60000。
[0089]
本发明中,所述聚酯s为无规共聚酯。
[0090]
本发明还提供了一种上文所述的聚酯组合物的制备方法,包括以下步骤:
[0091]
将聚乳酸和聚酯s混合后,经熔融挤出造粒,得到聚酯组合物。
[0092]
所述聚酯组合物的制备方法中,采用的原料组分同上,在此不再赘述。
[0093]
在本发明的某些实施例中,所述熔融的温度为170~250℃,时间为2~10min。在某些实施例中,所述熔融的温度为180℃或230℃,时间为3min或5min。
[0094]
在本发明的某些实施例中,所述熔融挤出造粒在双螺杆挤出机中进行。
[0095]
本发明还提供了一种上文所述的聚酯组合物作为阻隔包装材料或介电材料的应用;
[0096]
所述聚酯组合物为上文所述的聚酯组合物,或为上文所述的制备方法制得的聚酯组合物。
[0097]
本发明提供的聚酯组合物为可生物降解的聚酯组合物。
[0098]
本发明提供的聚酯组合物具有良好的韧性和阻隔性能。其断裂伸长率从纯pla的5%提高到了100%以上,无缺口冲击强度由纯pla的14kj/m2提高至35kj/m2以上;与纯pla相比,全生物基聚酯组合物的氧气透过率也大幅度降低,氧气透过系数低于150cm3/m2·
24h
·
0.1mpa,说明其阻隔性明显变好。与纯pla相比,全生物基聚酯组合物的拉伸强度也略有增加。同时,聚酯组合物的介电常数也相应增加。
[0099]
本发明对上文采用的原料来源并无特殊的限制,可以为一般市售。
[0100]
为了进一步说明本发明,以下结合实施例对本发明提供的一种聚酯组合物、其制备方法及应用进行详细描述,但不能将其理解为对本发明保护范围的限定。
[0101]
以下实施例中所用的试剂均为市售。
[0102]
实施例1
[0103]
聚酯s的制备:
[0104]
步骤1):在氮气保护下,将2,5-呋喃二甲酸、1,6-己二酸与1,4-丁二醇在催化剂钛酸四丁酯的作用下进行酯交换反应,180℃搅拌反应4h,反应压力为0.01mpa,生成预聚物;其中,1,4-丁二醇占1,6-己二酸和2,5-呋喃二甲酸的摩尔总和的摩尔比为3:1;2,5-呋喃二甲酸与1,6-己二酸的摩尔比为7:3;钛酸四丁酯占1,6-己二酸和2,5-呋喃二甲酸的摩尔总和的摩尔比为0.3%。
[0105]
步骤2):将步骤1)制备的预聚物,抽真空到5pa,230℃搅拌反应10h,即得聚酯s。
[0106]
对制得的聚酯s进行核磁氢谱分析,结果如图1所示。图1为本发明实施例1的聚酯s的核磁氢谱图。图1中既出现了呋喃环上的质子的化学位移a,又出现了己二酸中ch2的化学位移d和f,证明确实合成了目标共聚酯聚呋喃二甲酸-己二酸丁二醇酯。
[0107]
所述聚酯s具有式ⅰ所示结构,其中,r1具有式ⅱ所示结构,p=4;r2具有式iii-2所
示结构;m=42,n=98。
[0108]
全生物基聚酯组合物的制备:
[0109]
将75重量份聚乳酸(重均分子量为254300)和25重量份所述聚酯s一起加入双螺杆挤出机,在180℃下熔融3min,挤出造粒,即得全生物基聚酯组合物1#。
[0110]
实施例2
[0111]
聚酯s的制备:
[0112]
步骤1):在氮气保护下,将2,4-呋喃二甲酸、1,5-戊二酸与1,4-丁二醇在催化剂钛酸四丁酯的作用下进行酯交换反应,180℃搅拌反应4h,反应压力为0.01mpa,生成预聚物;其中,1,4-丁二醇占1,5-戊二酸和2,4-呋喃二甲酸的摩尔总和的摩尔比为3:1;2,4-呋喃二甲酸与1,5-戊二酸的摩尔比为3:3;钛酸四丁酯占1,5-戊二酸和2,4-呋喃二甲酸的摩尔总和的摩尔比为0.3%。
[0113]
步骤2):将步骤1)制备的预聚物,抽真空到5pa,230℃搅拌反应10h,即得聚酯s。
[0114]
所述聚酯s具有式ⅰ所示结构,其中,r1具有式ⅱ所示结构,p=3;r2具有式iii-1所示结构;m=118,n=118。
[0115]
全生物基聚酯组合物的制备:
[0116]
将60重量份聚乳酸(重均分子量为254300)和40重量份所述聚酯s一起加入双螺杆挤出机,在180℃下熔融5min,挤出造粒,即得全生物基聚酯组合物2#。
[0117]
实施例3
[0118]
聚酯s的制备:
[0119]
步骤1):在氮气保护下,将2,5-噻吩二甲酸、1,4-丁二酸与1,4-丁二醇在催化剂草酸亚锡的作用下进行酯交换反应,210℃搅拌反应4h,反应压力为0.01mpa,生成预聚物;其中,1,4-丁二醇占2,5-噻吩二甲酸和1,4-丁二酸的摩尔总和的摩尔比为3:1;2,5-噻吩二甲酸与1,4-丁二酸的摩尔比为6:4;草酸亚锡占2,5-噻吩二甲酸和1,4-丁二酸的摩尔总和的摩尔比为0.3%。
[0120]
步骤2):将步骤1)制备的预聚物,抽真空到5pa,230℃搅拌反应8h,即得聚酯s。
[0121]
所述聚酯s具有式ⅰ所示结构,其中,r1具有式ⅱ所示结构,p=2;r2具有式iii-3所示结构;m=56,n=84。
[0122]
全生物基聚酯组合物的制备:
[0123]
将68重量份聚乳酸(重均分子量为254300)和32重量份所述聚酯s一起加入双螺杆挤出机,在180℃下熔融5min,挤出造粒,即得全生物基聚酯组合物3#。
[0124]
实施例4
[0125]
聚酯s的制备:
[0126]
步骤1):在氮气保护下,将2,4-呋喃二甲酸、1,6-己二酸与1,4-丁二醇在催化剂草酸亚锡的作用下进行酯交换反应,180℃搅拌反应4h,反应压力为0.01mpa,生成预聚物;其中,1,4-丁二醇占1,6-己二酸和2,4-呋喃二甲酸的摩尔总和的摩尔比为3:1;2,4-呋喃二甲酸与1,6-己二酸的摩尔比为7:3;草酸亚锡占1,6-己二酸和2,4-呋喃二甲酸的摩尔总和的摩尔比为0.3%。
[0127]
步骤2):将步骤1)制备的预聚物,抽真空到5pa,230℃搅拌反应9h,即得聚酯s。
[0128]
所述聚酯s具有式ⅰ所示结构,其中,r1具有式ⅱ所示结构,p=2;r2具有式iii-1所
示结构;m=39,n=91。
[0129]
全生物基聚酯组合物的制备:
[0130]
将65重量份聚乳酸(重均分子量为254300)和35重量份所述聚酯s一起加入双螺杆挤出机,在180℃下熔融5min,挤出造粒,即得全生物基聚酯组合物4#。
[0131]
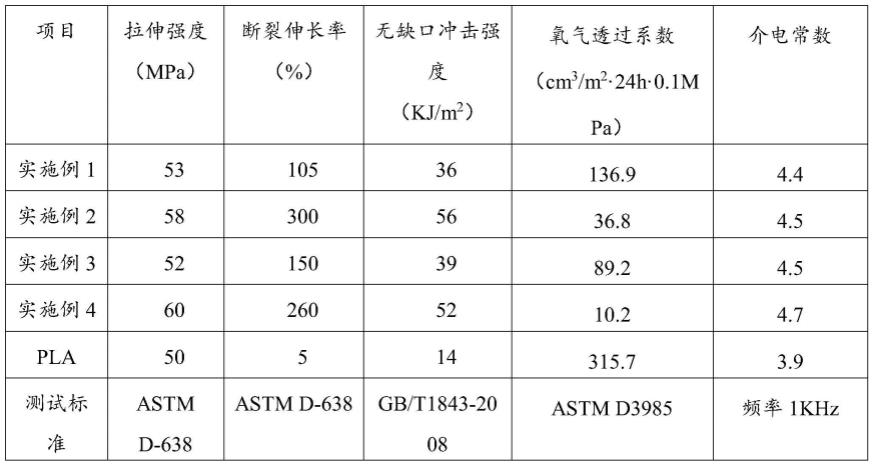
本发明对实施例1~4的全生物基聚酯组合物的性能进行检测,结果如表1所示。具体的,拉伸测试按照astm d638进行;无缺口冲击试验按照gb/t1843-2008进行;氧气阻隔性能按照astm d3985进行测定。
[0132]
表1实施例1~4的全生物基聚酯组合物的性能检测结果
[0133][0134]
从表1可知,本发明制备的全生物基聚酯组合物具有良好的韧性和阻隔性能。其断裂伸长率从纯pla的5%提高到了100%以上,无缺口冲击强度由纯pla的14kj/m2提高至35kj/m2以上;与纯pla相比,全生物基聚酯组合物的氧气透过率也大幅度降低,氧气透过系数低于150cm3/m2·
24h
·
0.1mpa,说明其阻隔性明显变好。与纯pla相比,全生物基聚酯组合物的拉伸强度也略有增加。同时,聚酯组合物的介电常数也相应增加。
[0135]
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。