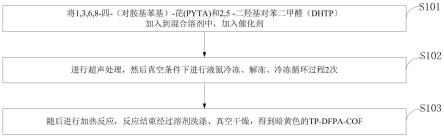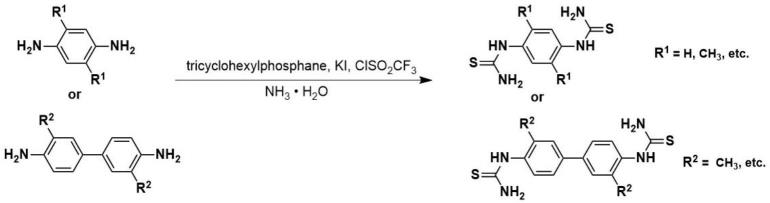
1.本发明属于cof单体合成技术领域,公开了一种苯基双硫脲类化合物的制备方法。
背景技术:
2.多种共价键连接方式如c-n、n-n、b-n、c-c及b-o等用来构建cof的骨架结构。而目前已报道的cofs材料中以硫脲结构连接的方式只有近一年的两例(h.l.qian,m.s.zhu,m.l.du.x.q.ran,x.p.yan,j.hazard.mater.,2022,427,128156;b.w.ma,c.z.li,l.zhang,l.p.zhai,f.j.hu,y.m.xu,h.j.qiao,z.wang,w.y.ai,l.w.mi,crystengcomm,2021,23,7576.),原因是构成硫脲cofs材料的硫脲单体合成的困难性。
3.我们知道苯基双硫脲化合物可以作为反应单体合成cofs材料。例如市面上1,4-苯双硫脲(cas:1519-70-6)的售卖比较便宜,其它取代基取代的苯基双硫脲化合物,很少有供应商提供,且价格昂贵。目前常用到的合成方法是用盐酸作为催化剂,1,4-苯二胺和异硫氰酸铵作为反应试剂,高温的条件下合成得到1,4-苯双硫脲(a.aumiiller,s.hiinig,liebigs ann.chem.,1986,1,142;u.hideaki,japan,1990,公开号:jp02042449a;j.thermoplast,compos.mater.,2016,29,312-326;s.habib,materials today:proceedings,2020,20,428,etc.)该方法虽然可以一步得到目标产物,但反应牵涉到高温和强酸,并不适合工业的放大应用。另外该方法得到类似结构的其它苯基双硫脲化合物存在收率低和难提纯的问题。因此通过该方法制备苯基双硫脲类化合物具有很大的局限性,不具有普适性。
4.也有文献报道通过1,4-苯二胺和二硫化碳作为起始原料,经过三步合成得到目标产物(b.w.ma,c.z.li,l.zhang,l.p.zhai,f.j.hu,y.m.xu,h.j.qiao,z.wang,w.y.ai,l.w.mi,crystengcomm,2021,23,7576.)。虽然该方法具有一定的底物普适性,但是反应步骤冗长,需要经过三步才能制备目标产物,且第二步收率只有10%左右。显然该方法明显不具备放大应用的能力。
5.通过上述分析,现有技术存在的问题及缺陷为:现有制备方法普适性差,收率低,反应条件苛刻及不适合放大生产,实用性差。
技术实现要素:
6.为克服相关技术中存在的问题,本发明公开实施例提供了一种苯基双硫脲类化合物的制备方法。
7.本发明目的在于提供一种反应条件温和,成本低、纯度和收率高、后处理操作简便,并具有较好工业化应用价值的苯基双硫脲类化合物的制备方法。
8.所述技术方案如下:一种苯基双硫脲类化合物的制备方法包括以下步骤:在有机溶剂中,将苯二胺化合物、有机膦化合物、碘化物混合均匀,氮气保护下,0℃下缓慢加入三氟甲磺酰氯后,在室温下进行反应1-4h。降至0℃,缓慢加入氨水后,搅拌5-30min后,升至温度50℃,继续反应0.5-2h后。将其离心分离、处理用的有机溶剂洗涤纯化、真空干燥,即得。
9.本发明的双硫脲化合物的制备方法,以苯基二胺类化合物为原料,反应过程温和且易控,通过一锅反应就可以得到产物,粗产品容易提纯且纯化操作简单。另外该方法普适性强,同样适用于类似结构的双硫脲化合物。产品的纯度高和收率高,使用的原料成本低,且可以放大生产,具有良好的工业应用价值。
10.在一个实施例中,所述的碘化物为:碘化钠、碘化钾、碘化胺、碘化锂、碘化铯和碘的一种,所述的有机膦化合物为:三苯基膦,三环己基膦、三(邻甲基苯基)膦、三(1-萘基)膦的一种。
11.在一个实施例中,苯基双硫脲类化合物的制备方法反应式为:
[0012][0013]
在一个实施例中,本发明提供的苯基双硫脲类化合物,其结构式如(1)式所示
[0014][0015]
在一个实施例中,为更好地降低副反应的发生,进一步提高反应选择性。优选的,所述溶剂为乙腈、dmf、dmso、thf,1,4-二氧六环、丙酮、乙酸乙酯中的至少一种。
[0016]
在一个实施例中,为更好地兼顾反应的平缓可控以及减小溶剂的用量。优选的,所述的苯二胺化合物:有机溶剂的物质的量比为1:(3-12)。
[0017]
在一个实施例中,为进一步促进原料的转化,减少副反应的发生。优选的,所述的苯二胺化合物:有机磷:碘化盐:三氟甲磺酰氯的物质的量之比为1:(2-6):(2-4):(2-4)。
[0018]
在一个实施例中,为更好地降低副反应的发生,进一步提高反应选择性。优选的,所述的反应步骤中加入氨水后,反应温度应控制在30-50℃。
[0019]
在一个实施例中,为进一步简化后处理操作及降低后处理成本。优选的,后处理用的洗涤溶剂为乙腈、thf、乙酸乙酯、乙醇和水的一种或几种。
[0020]
本发明的另一目的在于合成得到的苯基双硫脲化合物可以作为共价有机框架材料的合成单体,与各种单体搭配,合成各种含有硫脲基团的共价有机框架材料。进而拓展了cofs材料的结构多样性,促进各类硫脲cofs材料在气体存储与吸附、分子识别与分离、智能传感、光电、能量储存、生物医药和催化等领域的应用性。
[0021]
结合上述的所有技术方案,本发明所具备的优点及积极效果为:
[0022]
第一、本发明提供了合成苯基双硫脲类化合物的一种新工艺,本发明提供的苯基双硫脲类化合物的合成方法与现有相比具有很大的优势,成本低,收率高,且适合工业放大。本发明提供的苯基双硫脲类化合物其中的一种应用是:可以作为cof材料的合成单体,
为cof的合成及应用提供了思路。
[0023]
第二、现有方法合成苯基双硫脲类化合物具有一定局限性,反应往往牵涉到高温和强酸,并不适合工业的放大应用。另外现有方法得到类似结构的其它苯基双硫脲化合物普遍存在收率低和难提纯的问题,致使提纯后处理工艺复杂,成本高,不适合工业应用。本发明开发一种简单快捷、温和、高效、能放大应用的方法显然很有科学意义和应用价值。
[0024]
第三、本发明的苯基双硫脲类化合物的制备方法通过一锅反应制备,过程温和易控,产品的纯度和收率高,底物普适性强,后处理简单快捷,且可以放大规模制备产物。这些优点显著降低了苯基双硫脲类化合物的制备成本,具有良好的工业应用价值。
[0025]
第四、本发明技术方案转化后具有比较大的商业价值,本发明的生产工艺操作简单可行,原料及工艺成本低,收率高,可以放大生产且收率可以保持。产物在市面上可以作为原料中间体,具有很好的市场前景。因而该发明具有比较大的工业应用价值和商业价值。
[0026]
第五、本发明的技术方案解决了以往苯基双硫脲类化合物的制备成本高,收率低,纯化工序复杂困难,工艺条件苛刻以及不容易放大生产的缺点。为人们设计合成其它硫脲类化合物提供了良好的科学思路。
附图说明
[0027]
此处的附图被并入说明书中并构成本说明书的一部分,示出了符合本公开的实施例,并与说明书一起用于解释本公开的原理。
[0028]
图1是本发明实施例提供的苯基双硫脲类化合物的制备方法流程图;
[0029]
图2是本发明实施例提供的1,4-苯二双硫脲化合物的核磁共振氢图;
[0030]
图3是本发明实施例提供的1,4-苯二双硫脲化合物的核磁共振碳图;
[0031]
图4是本发明实施例提供的是2,5-二甲基-1,4-苯双硫脲化合物的核磁共振氢图;
[0032]
图5是本发明实施例提供的2,5-二甲基-1,4-苯双硫脲化合物的核磁共振碳图;
[0033]
图6是本发明实施例提供的3,3'-二甲基-联苯双硫脲化合物的核磁共振氢图;
[0034]
图7是本发明实施例提供的3,3'-二甲基-联苯双硫脲化合物的核磁共振碳图;
[0035]
图8是本发明应用实施例1提供的反应式和材料的小角pxrd图;
[0036]
图9是本发明应用实施例2提供的反应式和材料的小角pxrd图;
[0037]
图10是本发明应用实施例3提供的反应式和材料的小角pxrd图。
具体实施方式
[0038]
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其他方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。
[0039]
如图1所示,本发明提供一种苯基双硫脲类化合物的制备方法包括以下步骤:
[0040]
s101,在有机溶剂中,将苯二胺化合物、有机膦化合物、碘化物混合均匀,氮气保护下,0℃下缓慢加入三氟甲磺酰氯后,在室温下进行反应1-4h。
[0041]
s102,降至0℃,缓慢加入氨水后,搅拌5-30min后,升至温度50℃,继续反应0.5-2h。
[0042]
s103,将其离心分离、处理用的有机溶剂洗涤纯化、真空干燥,即得。
[0043]
在一优选实施例中,所述的碘化物为:碘化钠、碘化钾、碘化胺、碘化锂、碘化铯和碘的一种,所述的有机膦化合物为:三苯基膦,三环己基膦、三(邻甲基苯基)膦、三(1-萘基)膦的一种。
[0044]
在一优选实施例中,苯基双硫脲类化合物的制备方法反应式为:
[0045][0046]
本发明还提供的苯基双硫脲类化合物,其结构式如(1)式所示
[0047][0048]
在一优选实施例中,所述溶剂为乙腈、dmf、dmso、thf,1,4-二氧六环、丙酮、乙酸乙酯中的至少一种。
[0049]
在一优选实施例中,所述的苯二胺化合物:有机溶剂的物质的量比为1:(3-12)。
[0050]
在一优选实施例中,所述的苯二胺化合物:有机磷:碘化盐:三氟甲磺酰氯的物质的量之比为1:(2-6):(2-4):(2-4)。
[0051]
在一优选实施例中,所述的反应步骤中加入氨水后,反应温度应控制在30-50℃。
[0052]
在一优选实施例中,后处理用到的洗涤溶剂为乙腈、thf、乙酸乙酯、乙醇和水的一种或几种。
[0053]
下面结合具体实施例对本发明的技术方案作进一步描述。
[0054]
实施例1
[0055]
本发明的1,4-苯二双硫脲化合物的制备方法实施例1,合成路线如下所示:
[0056][0057]
合成步骤:在50ml dmf中,将10mmol的1,4-苯二胺、25mmol三环己基膦、24mmol碘化钾混合均匀,氮气保护下,0℃下缓慢加入24mmol三氟甲磺酰氯后,在室温下进行反应3h。降至0℃,缓慢加入氨水后,搅拌10min后,升至温度50℃,继续反应1h后。将其离心分离、thf和水洗涤纯化、真空干燥,即得白色固体产物1.94克,收率86%。
[0058]
产品的核磁共振氢谱和碳谱如图2和图3所示,表征数据为:1h nmr(400mhz,氘代dmso)δ9.61(s,2h),7.30(s,8h);
13
c nmr(100mhz,氘代dmso)δ181.5,136.0,124.2。
[0059]
实施例2
[0060]
本实施例与实施例1的区别在于:反应采用的溶剂为乙腈,有机膦为三苯基膦,收率为56%。
[0061]
本实施例中其他步骤与实施例1相同,在此不再赘述。
[0062]
实施例3
[0063]
本实施例与实施例1的区别在于:0℃下缓慢加入氨水后,搅拌20min后,升至温度40℃,收率78%。
[0064]
本实施例中其他步骤与实施例1相同,在此不再赘述。
[0065]
实施例4
[0066]
本实施例与实施例1的区别在于后处理洗涤溶剂为乙腈,收率为82%。
[0067]
本实施例中其他步骤与实施例1相同,在此不再赘述。
[0068]
实施例5
[0069]
百克级合成步骤:在1.5l dmf中,将0.5mol的1,4-苯二胺、1.25mol三环己基膦、1.2mol碘化钾混合均匀,氮气保护下,0℃下缓慢加入1.2mol三氟甲磺酰氯后,在室温下进行反应3h。降至0℃,缓慢加入氨水后,搅拌10min后,升至温度50℃,继续反应1h后。将其离心分离、thf和水洗涤纯化、真空干燥,即得白色固体产物101克,收率89%。
[0070]
在本发明的1,4-苯双硫脲的制备方法的其他实施例中,反应原料的用量、反应温度、反应时间、溶剂的量和溶剂种类及其它具体反应条件可以在本发明限定的范围内进行适应性调整,可达到与实施例相当的效果。
[0071]
本发明的2,5-二甲基-1,4-苯双硫脲化合物的制备方法实施例6,合成路线如下所示:
[0072][0073]
合成步骤:在50ml dmf中,将10mmol的2,5-二甲基-1,4-苯二胺、25mmol三环己基膦、24mmol碘化钾混合均匀,氮气保护下,0℃下缓慢加入24mmol三氟甲磺酰氯后,在室温下进行反应3h。降至0℃,缓慢加入氨水后,搅拌10min后,升至温度40℃,继续反应1h后。将其离心分离、thf和水洗涤纯化、真空干燥,即得白色固体产物2.24克,收率88%。
[0074]
产品的核磁共振氢谱和碳谱如图4和图5所示,表征数据为:1h nmr(600mhz,氘代dmso)δ9.29(s,2h),7.71(s,2h),7.01(s,2h),6.51(s,2h),2.10(s,6h);
13
c nmr(150mhz,氘代dmso)δ181.7,135.7,133.6,130.2,17.4。
[0075]
实施例7
[0076]
本实施例与实施例6的区别在于:反应采用的溶剂为1,4-二氧六环,有机膦试剂为三苯基膦,收率为65%。
[0077]
本实施例中其他步骤与实施例6相同,在此不再赘述。
[0078]
实施例8
[0079]
本实施例与实施例6的区别在于:0℃下缓慢加入氨水后,搅拌20min后,升至温度50℃,收率82%。
[0080]
本实施例中其他步骤与实施例6相同,在此不再赘述。
[0081]
实施例9
[0082]
本实施例与实施例6的区别在于后处理洗涤溶剂为乙酸乙酯,收率为81%。
[0083]
本实施例中其他步骤与实施例6相同,在此不再赘述。
[0084]
实施例10
[0085]
百克级合成步骤:在1.5l dmf中,将0.5mol的2,5-二甲基-1,4-苯二胺、1.25mol三环己基膦、1.2mol碘化钾混合均匀,氮气保护下,0℃下缓慢加入1.2mol三氟甲磺酰氯后,在室温下进行反应3h。降至0℃,缓慢加入氨水后,搅拌10min后,升至温度40℃,继续反应1h后。将其离心分离、thf和水洗涤纯化、真空干燥,即得白色固体产物116克,收率91%。
[0086]
在本发明的2,5-二甲基-1,4-苯双硫脲的制备方法的其他实施例中,反应原料的用量、反应温度、反应时间、溶剂的量和溶剂种类及其它具体反应条件可以在本发明限定的范围内进行适应性调整,可达到与实施例相当的效果。
[0087]
本发明的3,3'-二甲基-联苯双硫脲化合物化合物的制备方法实施例11,合成路线如下所示:
[0088][0089]
合成步骤:在50ml dmf中,将10mmol的3,3'-二甲基-联苯二胺、25mmol三环己基膦、24mmol碘化钾混合均匀,氮气保护下,0℃下缓慢加入24mmol三氟甲磺酰氯后,在室温下进行反应3h。降至0℃,缓慢加入氨水后,搅拌10min后,升至温度40℃,继续反应1h后。将其离心分离、thf和水洗涤纯化、真空干燥,即得白色固体产物2.97克,收率90%。
[0090]
产品的核磁共振氢谱和碳谱如图6和图7所示,表征数据为:1h nmr(600mhz,dmso)δ9.23(s,2h),7.69(s,2h),7.54(d,j=1.8hz,2h),7.47(dd,j=8.4,1.8hz,2h),7.31(d,j=8.4hz,2h),7.03(s,2h),2.26(s,6h);
13
c nmr(150mhz,dmso)δ182.1,138.1,137.0,135.2,129.1,128.3,124.9,18.3。
[0091]
实施例12
[0092]
本实施例与实施例11的区别在于:添加剂碘化物采用的是碘化钠,收率为81%。
[0093]
本实施例中其他步骤与实施例11相同,在此不再赘述。
[0094]
实施例13
[0095]
本实施例与实施例11的区别在于:缓慢加入氨水后,搅拌20min后,升至温度40℃,继续反应2h,收率86%。
[0096]
本实施例中其他步骤与实施例11相同,在此不再赘述。
[0097]
实施例14
[0098]
本实施例与实施例11的区别在于后处理洗涤溶剂为乙腈,收率为83%。
[0099]
本实施例中其他步骤与实施例11相同,在此不再赘述。
[0100]
实施例15
[0101]
百克级合成步骤:在1.5l dmf中,将0.5mol的3,3'-二甲基-联苯二胺、1.25mol三环己基膦、1.2mol碘化钾混合均匀,氮气保护下,0℃下缓慢加入1.2mol三氟甲磺酰氯后,在室温下进行反应3h。降至0℃,缓慢加入氨水后,搅拌10min后,升至温度40℃,继续反应1h
后。将其离心分离、thf和水洗涤纯化、真空干燥,即得白色固体产物145克,收率88%。
[0102]
在本发明的3,3'-二甲基-联苯双硫脲的制备方法的其他实施例中,反应原料的用量、反应温度、反应时间、溶剂的量和溶剂种类及其它具体反应条件可以在本发明限定的范围内进行适应性调整,可达到与实施例相当的效果。
[0103]
为了证明本技术发明的应用性。已经将本发明涉及到的三种苯基双硫脲化合物作为单体应用到cof材料的合成中。以三醛基间苯三酚和这三种苯基双硫脲化合物反应制备cof材料。具体材料合成过程如下:
[0104]
应用实施例1
[0105]
tp-tu-cof材料的合成:取一10毫升的安培瓶,称量20mg三醛基间苯三酚和32mg 1,4-苯二双硫脲,加入0.6ml nmp,0.4mltcb和0.1ml的6m醋酸水溶液。将其置于真空条件,77k温度下循环进行液氮冷冻、解冻、冷冻过程2次。然后密封,置于120℃下反应三天,就可以得到tp-tu-cof材料。反应式和材料的小角pxrd如下图8所示,其中pxrd图中的4
°‑
30
°
的衍射峰证实了成功合成tp-tu-cof材料。
[0106]
应用实施例2
[0107]
tp-dmtu-cof材料的合成:取一10毫升的安培瓶,称量20mg三醛基间苯三酚和36mg 2,5-二甲基-1,4-苯双硫脲,加入0.6ml nmp,0.4mltcb和0.1ml的6m醋酸水溶液。将其置于真空条件,77k温度下循环进行液氮冷冻、解冻、冷冻过程2次。然后密封,置于150℃下反应三天,就可以得到tp-tu-cof材料。反应式和材料的小角pxrd如下图9所示,其中pxrd图中的4
°‑
30
°
的特征衍射峰证实了成功合成tp-dmtu-cof材料。
[0108]
应用实施例3
[0109]
tp-dmptu-cof材料的合成:取一10毫升的安培瓶,称量20mg三醛基间苯三酚和36mg 3,3'-二甲基-联苯双硫脲,加入0.6ml nmp,0.4mltcb和0.1ml的6m醋酸水溶液。将其置于真空条件,77k温度下循环进行液氮冷冻、解冻、冷冻过程2次。然后密封,置于150℃下反应三天,就可以得到tp-tu-cof材料。反应式和材料的小角pxrd如下图10所示,其中pxrd图中的3.0
°‑
20
°
的特征衍射峰证实了成功合成tp-dmptu-cof材料。
[0110]
以上所述,仅为本发明较优的具体的实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。