一种冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚、制备方法及其应用
技术领域
1.本发明属于有机合成领域,尤其涉及一种冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚、制备方法及其应用。
背景技术:
2.新型手性催化剂的设计与合成在不对称催化反应的研究中起着非常关键的作用,手性催化剂的空间结构或电子性质的变化显著影响其催化性能,探讨催化剂的手性、产物的旋光性及对映选择性之间的关系对于指导不对称合成具有重要的意义。
3.手性1,1
′‑
联
‑
2,2
′‑
萘酚(binol)是一类重要的轴手性化合物,现已被广泛地用作手性配体或手性催化剂,在多种不对称催化反应中具有高对映选择性。binol的结构中的两个萘环体积大,因此空间位阻大而具有刚性,又因两环之间有共轴效应,仍有一定旋转余地而具有柔性,其3,3
′‑
位引入取代基后往往可以在不对称催化反应中取得更好的立体选择性,常用的3,3
′‑
位取代基为刚性的芳基或取代芳基。
4.公开号为cn108658774b的中国专利文献中公开了一种新型手性硝基联萘酚及其衍生物的合成方法,该发明以手性联萘酚为原料,与绿色硝化试剂叔丁基亚硝酸酯(tbn)在室温条件下,以四氢呋喃作为反应溶剂,发生自由基硝化反应,获得手性硝基联萘酚,实现了绿色硝化。
5.公开号为cn102936219b的中国专利文献中公开了一种手性6,6
′‑
二咔唑基联萘酚,制备方法包括:在碱存在下,钯催化s构型6,6
′‑
二溴联萘酚和咔唑的c-n偶联得到s构型6,6
′‑
二咔唑基联萘酚。该手性6,6
′‑
二咔唑基联萘酚可用作不对称催化的手性配体或用于制备蓝色有机发光二极管。
6.公开号为cn109810129a的中国专利文献中公开了一种联萘偶氮苯环状光敏手性分子及其制备方法及用途,制备方法包括:将6,6
′
二溴
‑
1,1
′‑
联
‑2‑
萘酚、碳酸钾、碘化钾及2
‑
(2
‑
氯乙氧基)乙醇,溶解于n,n
‑
二甲基甲酰胺中,得中间体1,与4
‑
戊氧基联苯硼酸溶解于1,4
‑
二氧六环中,与碳酸钾水溶液和四三苯基膦钯反应得中间体2,经二氯甲烷溶解,加入三乙胺、4
‑
二甲氨基吡啶、对甲苯磺酰氯,反应得中间体3;再与2,2
′‑
二羟基偶氮苯和二苯并
‑
18
‑
冠醚
‑
6于在氮气氛围下,加入碳酸铯反应,得目标产物t。该发明的联萘偶氮苯环状光敏手性分子可通过光致顺反异构来实现对胆甾相液晶螺距的调控,与液晶分子间有较好的相容性。
技术实现要素:
7.本发明提供了一种冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚,具有大立体位阻,且富电子冠醚的引入提高了其催化反应活性和立体选择性,可作为催化剂应用于不对称催化反应。
8.具体采用的技术方案如下:
9.一种冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚,具有如式(ⅰ)所示的结构式:
[0010][0011]
其中,式(ⅰ)中,n为1~5中的整数,ar为亚芳基或取代亚芳基;所述的亚芳基为二价苯基、二价联苯基、二价萘基中的任一种;所述的取代亚芳基的取代基包括氟原子、氯原子、烷基、芳基、烷氧基、芳氧基、烃硫基、氰基、硝基、乙烯基、取代乙烯基或炔基。
[0012]
冠醚是一类高度富电子的大环主体,常用于对金属阳离子和有机阳离子的主客体络合。本发明在手性1,1
′‑
联
‑
2,2
′‑
萘酚中引入大立体位阻且富电子的冠醚,可以使得催化活性中心1,1
′‑
联
‑
2,2
′‑
萘酚位于一个手性诱导效果更佳、四周电子云密度较高的空腔中,因而有望在不对称催化反应中获得比一般的手性1,1
′‑
联
‑
2,2
′‑
萘酚更好的催化效果。
[0013]
优选的,所述的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚的结构式为:
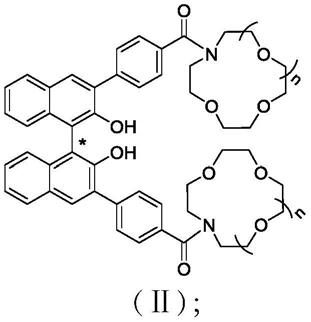
[0014][0015]
其中,式(ⅱ)中,n为1~5中的整数。
[0016]
本发明还提供了所述的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚的制备方法,包括以下步骤:
[0017]
(1)手性1,1
′‑
联
‑
2,2
′‑
萘酚由甲氧基甲基醚保护羟基得到手性2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘,手性2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘在丁基锂的存在下与碘反应得到中间产物a,所述的手性2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘与碘的摩尔比为1:2~4;
[0018]
(2)将中间产物a与芳基硼酸在钯催化剂、碱存在下进行suzuki偶联反应得到中间产物b,所述的中间产物a与芳基硼酸的摩尔比为1:2~4;
[0019]
(3)使中间产物b在碱性条件下发生水解反应得到中间产物c;
[0020]
(4)将中间产物c与氮杂冠醚进行酰胺化反应得到中间产物d,所述的中间产物c与氮杂冠醚的摩尔比为1:2~3;
[0021]
(5)中间产物d发生脱保护反应得到所述的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚。
[0022]
所述的手性1,1
′‑
联
‑
2,2
′‑
萘酚包括(s)
‑
1,1
′‑
联
‑
2,2
′‑
萘酚和(r)
‑
1,1
′‑
联
‑
2,2
′‑
萘酚,因此,所述的手性2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘也包括(s)
‑
2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘和(r)
‑
2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘。
[0023]
所述的芳基硼酸的结构式为式(ⅲ)所示结构式的任一种:
[0024][0025]
其中r为氢原子、氟原子、氯原子、烷基、芳基、烷氧基、芳氧基、烃硫基、氰基、硝基、乙烯基、取代乙烯基或炔基。
[0026]
所述的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚具有手性,可以是一种对映异构体,可以是外消旋体,可以是对映异构体的非消旋混合物。
[0027]
当所述的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚的光学纯度≥30%ee时,可以将非手性反应物转变为手性产物。
[0028]
本发明还提供了所述的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚作为催化剂在不对称催化反应中的应用,所述的不对称催化反应包括aldol反应、michael加成反应和diels
‑
alder等环加成反应。
[0029]
1,1
′‑
联
‑
2,2
′‑
萘酚的3,3
′‑
位修饰基团通常为大立体位阻的芳基等刚性基团,冠醚也具有大立体位阻,但它同时具有柔性,冠醚的引入使得所述的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚作为催化剂在催化反应过程中能够根据反应底物结构的不同而适当调整自身的空间结构,来满足催化剂与底物的自适应;同时,冠醚上多个氧原子的存在使得催化剂会与底物形成更多的氢键等超分子作用;此外,引入冠醚还有可能通过冠醚与金属阳离子或有机阳离子的主客体络合来实现对催化剂空间结构和电子性质的调控。
[0030]
优选的,所述的不对称催化反应为烯基硼酸与α,β
‑
不饱和酮的michael加成反应;反应过程为:α,β
‑
不饱和酮与烯基硼酸在所述的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚作为催化剂的条件下,混合添加剂金属盐、分子筛(ms)和有机溶剂,在氮气保护下,50~140℃反应24~72h。
[0031]
进一步优选的,所述的α,β
‑
不饱和酮为4
‑
苯基
‑3‑
丁烯
‑2‑
酮、3
‑
戊烯
‑2‑
酮、3
‑
庚烯
‑2‑
酮、3
‑
壬烯
‑2‑
酮、查耳酮、1
‑
(4
‑
甲氧基苯基)
‑3‑
苯基丙
‑2‑
烯
‑1‑
酮、4
′‑
氟查耳酮、4
‑
甲氧基查耳酮、4
‑
氯查耳酮、4
‑
甲氧基苯亚甲基丙酮、4
‑
氯苯亚甲基丙酮、1
‑
[3
‑
(三氟甲基)苯基]丁
‑1‑
烯
‑3‑
酮、2,4
‑
二氯苯亚甲基丙酮、4
‑
(4
‑
羟基
‑3‑
甲氧苯基)
‑3‑
丁烯
‑2‑
酮、异丁基苯乙烯酮;所述的烯基硼酸为反式
‑2‑
苯基乙烯基硼酸;所述的添加剂金属盐为mg
(otbu)2、mgcl2或mg(oet)2;所述的有机溶剂为二氯甲烷、二氯乙烷、甲苯、二甲苯;所述的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚的光学纯度≥85%ee。
[0032]
进一步优选的,以α,β
‑
不饱和酮的摩尔数计,α,β
‑
不饱和酮与烯基硼酸的加入量摩尔比为1:1.2,催化剂的加入量为α,β
‑
不饱和酮加入量的10
‑
20%,分子筛加入量为α,β
‑
不饱和酮加入量的5
‑
10mg/0.01mmol;添加剂金属盐的加入量为α,β
‑
不饱和酮加入量的10
‑
20%;α,β
‑
不饱和酮在反应体系中的浓度为0.02
‑
0.2m。
[0033]
与现有技术相比,本发明的优异效果在于:
[0034]
(1)本发明公开的一种冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚及其合成方法,具有大立体位阻和柔性,引入富电子冠醚提高了催化反应活性和立体选择性,合成方法可控,产率较高。
[0035]
(2)本发明通过在手性1,1
′‑
联
‑
2,2
′‑
萘酚的3,3
′‑
位引入大环冠醚这种超分子基元,使得利用氢键、主客体络合等超分子作用来调控催化剂空间结构和电子性质成为可能。
[0036]
(3)本发明公开的一种冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚可作为催化剂应用于烯基硼酸与α,β
‑
不饱和酮的不对称迈克尔加成反应中,手性产物产率可达到90%,产物的光学纯度可达到99%;此外,反应过程中无需采用贵金属、不稳定的硼酸酯等,操作简便、方法可控。
附图说明
[0037]
图1为实施例1中间产物a的核磁共振氢谱图。
[0038]
图2为实施例1中间产物a的核磁共振碳谱图。
[0039]
图3为实施例1中间产物b的核磁共振氢谱图。
[0040]
图4为实施例1中间产物b的核磁共振碳谱图。
[0041]
图5为实施例1中间产物c的核磁共振氢谱图。
[0042]
图6为实施例1中间产物c的核磁共振碳谱图。
[0043]
图7为实施例1中间产物d的核磁共振氢谱图。
[0044]
图8为实施例1中间产物d的核磁共振碳谱图。
[0045]
图9为实施例1冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
1的核磁共振氢谱图。
[0046]
图10为实施例1冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
1的核磁共振碳谱图。
具体实施方式
[0047]
下面结合实施例,进一步阐明本发明。应理解,这些实施例仅用于说明本发明,而不用于限制本发明的范围。
[0048]
实施例1
‑
3中,冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚的结构式如式(ⅱ)所示,其中,n为1~5;
[0049]
上述冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚的合成路线图如式(ⅳ)所示,其中,n为1~5;
[0050][0051]
应用例1
‑
23中,以实施例中制得的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚为催化剂催化α,β
‑
不饱和酮与反式
‑2‑
苯基乙烯基硼酸的加成反应,反应路线图如下式(
ⅴ
)所示:
[0052][0053]
实施例1
[0054]
(1)中间产物a的制备:将(s)
‑
2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘(17.4mmol)加入到干燥的三颈烧瓶中,氮气保护下加入重蒸的四氢呋喃(100ml),搅拌15分钟后将20.8ml 2.5m正丁基锂的正己烷溶液缓慢加入到(s)
‑
2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘的四氢呋喃0℃溶液中。滴加完成后继续搅拌15分钟,缓慢升到25℃反应3小时。将反应液降至
‑
78℃,再将溶解在四氢呋喃(30ml)中的碘(52.0mmol)滴加到上述溶液中,滴加完成后升温到室温,在室温下搅拌3小时,点板跟踪反应。反应结束后,用甲醇淬灭反应,随后加入饱和硫代硫酸钠水溶液,用乙酸乙酯萃取至水中不再含有产物。合并有机相,用饱和食盐水洗涤后,浓缩有机相,残余物通过硅胶柱层析(石油醚:乙酸乙酯=200:1)得到中间产物a(白色固体,7.80g,产率为72%)。
[0055]
(2)中间产物b的制备:将中间产物a(3.19mmol)、4
‑
甲氧酰基苯硼酸(6.54mmol)、磷酸三钾(19.2mmol)、三(二亚苄基丙酮)二钯(0.064mmol)和三环己基膦(0.146mmol)加入到100ml反应瓶中,氮气保护下加入1,4
‑
二氧六环(40ml)和水(10ml)作为反应溶剂,100℃下加热回流反应18小时。冷却到室温后,加入饱和edta溶液淬灭反应,用二氯甲烷萃取至水相中不再含有产物。合并有机相,用饱和食盐水洗涤后,浓缩有机相,残余物通过硅胶柱层析(石油醚:乙酸乙酯=10:1)得到中间产物b(白色固体,1.25g,产率为61%)。
[0056]
(3)中间产物c的制备:将中间产物b(1.55mmol)加入100ml反应瓶中,氮气保护下加入四氢呋喃(30ml)和10%naoh水溶液(5ml),65℃下加热回流12小时,点板跟踪反应。反应完全后,冷却至0℃,滴加入4m hcl调节ph至3~4,用乙酸乙酯萃取直至水相中无产物。合并有机相,无水硫酸钠干燥,旋蒸除去溶剂后干燥得到中间产物c(白色固体,1.79g,产率为94%)。
[0057]
(4)中间产物d的制备:将中间产物c(0.813mmol)、1
‑
氮杂
‑
15
‑
冠
‑
5醚(1.71mmol)、4
‑
(二甲氨基)吡啶(1.87mmol)、n
‑
(3
‑
二甲氨基丙基)
‑
n
′‑
乙基碳二亚胺盐酸盐(1.79mmol)加入到100ml反应瓶中,在氮气保护下加入25ml二氯甲烷,室温反应24小时,点板跟踪反应。反应结束后反应液水洗,再用二氯甲烷萃取水相至水相中不再含有产物,合并有机相,用饱和食盐水洗涤后,浓缩有机相,残余物通过硅胶柱层析(先用乙酸乙酯,再用二氯甲烷:甲醇=60:1)得到中间产物d(白色固体,458mg,产率为55%)。
[0058]
(5)冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚的合成:将中间产物d(0.197mmol)加入50ml反应瓶中,加入cah2重蒸的二氯甲烷(20ml),搅拌5分钟后再加入三氟乙酸(0.10ml),25℃室温下反应12h,点板跟踪反应。反应结束后,用50ml水洗,水相用二氯甲烷再萃取三次。有机相合并后用饱和食盐水洗涤,再用无水硫酸钠干燥。浓缩溶剂后,残余物通过硅胶柱层析(二氯甲烷:甲醇=50:1),得到冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
1(白色固体,146mg,产率为80%)。
[0059]
中间产物a的核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示,证明了中间产物a的成功合成,中间产物a的结构式如下所示:
[0060][0061]
中间产物b的核磁共振氢谱图如图3所示,核磁共振碳谱图如图4所示,证明了中间产物b的成功合成,中间产物b的结构式如下所示:
[0062][0063]
中间产物c的核磁共振氢谱图如图5所示,核磁共振碳谱图如图6所示,证明了中间产物c的成功合成,中间产物c的结构式如下所示:
[0064][0065]
中间产物d的核磁共振氢谱图如图7所示,核磁共振碳谱图如图8所示,证明了中间产物d的成功合成,中间产物d的结构式如下所示:
[0066][0067]
本实施例中冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
1的核磁共振氢谱图如图9所示,核磁共振碳谱图如图10所示,证明了冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
1的成功合成。
[0068]
实施例2
[0069]
本实施例与实施例1的制备方法和参数相同,区别仅在于将氮杂冠醚换为1
‑
氮杂
‑
18
‑
冠
‑
6醚(1.71mmol),得到冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
2(白色固体,477mg,产率为53%)。
[0070]
实施例3
[0071]
本实施例与实施例1的制备方法和参数相同,区别仅在于将手性2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘换为(r)
‑
2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘,得到冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
3。
[0072]
其中,步骤(1)得到7.50g中间产物a(白色固体),产率为69%;步骤(2)得到1.36g中间产物b(白色固体),产率为66%;步骤(3)得到1.83g中间产物c(白色固体),产率为96%;步骤(4)得到423mg中间产物d(白色固体),产率为51%;步骤(5)得到155mg冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
3(白色固体),产率为85%。
[0073]
实施例4
[0074]
本实施例与实施例1反应参数和条件相同,区别仅在于芳基硼酸换为4
′‑
(甲氧酰基)联苯
‑4‑
硼酸,得到冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
4。
[0075]
实施例5
[0076]
本实施例与实施例1反应参数和条件相同,区别仅在于芳基硼酸换为4
‑
(甲氧酰基)萘
‑1‑
硼酸,得到冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
5。
[0077]
实施例6
[0078]
本实施例与实施例1反应参数和条件相同,区别仅在于芳基硼酸换为2
‑
氯
‑4‑
(甲氧酰基)苯硼酸,得到冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
6。
[0079]
实施例7
[0080]
本实施例与实施例1反应参数和条件相同,区别仅在于芳基硼酸换为3
‑
甲氧基
‑4‑
甲氧酰基苯硼酸,得到冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
7。
[0081]
实施例8
[0082]
本实施例与实施例1反应参数和条件相同,区别仅在于芳基硼酸换为2
‑
(甲氧酰基)苯硼酸,得到冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
8。
[0083]
实施例9
[0084]
本实施例与实施例1反应参数和条件相同,区别仅在于所述的手性2,2
′‑
二(甲氧基甲氧基)
‑
1,1
′‑
联二萘与碘的摩尔比为1:3;所述的中间产物a与4
‑
甲氧酰基苯硼酸的摩尔比为1:3;所述的中间产物c与1
‑
氮杂
‑
15
‑
冠
‑
5醚的摩尔比为1:3,得到冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
9。
[0085]
应用例1
[0086]
以实施例1中的冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚m
‑
1为催化剂催化4
‑
苯基
‑3‑
丁烯
‑2‑
酮与反式
‑2‑
苯基乙烯基硼酸的加成反应;
[0087]
将4
‑
苯基
‑3‑
丁烯
‑2‑
酮(0.040mmol)、反式
‑2‑
苯基乙烯基硼酸(0.048mmol)、mg(otbu)2(0.0040mmol)、m
‑
1(0.0060mmol)和分子筛(20mg)加入到干燥的反应瓶中。在氮气保护下,加入重蒸的甲苯0.8ml,在70℃下反应48小时。反应结束后,冷却至室温,用甲醇淬灭反应。过滤后旋干反应液,通过核磁共振氢谱确定反应转化率;浓缩残余物通过柱层析分离加成产物(石油醚:二氯甲烷为1~2:1);采用手性高效液相色谱测定加成产物的光学纯度。
[0088]
本应用例中,反应转化率为42%;加成产物的光学纯度为89%ee。
[0089]
应用例2
[0090]
本应用例与应用例1反应参数相同,区别仅在于添加剂金属盐换为mgcl2,mgcl2的加入量换为0.0080mmol。反应转化率为40%;加成产物的光学纯度为89%ee。
[0091]
应用例3
[0092]
本应用例与应用例1反应参数相同,区别仅在于添加剂金属盐换为mg(oet)2。反应转化率为40%;加成产物的光学纯度为80%ee。
[0093]
应用例4
[0094]
本应用例与应用例1反应参数相同,区别在于反应温度为110℃。反应转化率为48%;加成产物的光学纯度为89%ee。
[0095]
应用例5
[0096]
本应用例与应用例1反应参数相同,区别在于4
‑
苯基
‑3‑
丁烯
‑2‑
酮的在反应体系中的浓度换为0.08m。反应转化率为39%;加成产物的光学纯度为83%ee。
[0097]
应用例6
[0098]
本应用例与应用例1反应参数相同,区别在于分子筛的用量为40mg。反应转化率为37%;加成产物的光学纯度为86%ee。
[0099]
应用例7
[0100]
本应用例与应用例4反应参数相同,区别在于催化剂m
‑
1的用量为0.008mmol。反应转化率为53%;加成产物的光学纯度为89%ee。
[0101]
应用例8
[0102]
本应用例与应用例4反应参数相同,区别在于催化剂选用实施例2中的m
‑
2。反应转化率为37%;加成产物的光学纯度为80%ee。
[0103]
应用例9
[0104]
本应用例与应用例1反应参数相同,区别在于有机溶剂选用二氯甲烷。反应转化率为35%;加成产物的光学纯度为78%ee。
[0105]
应用例10
[0106]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为3
‑
戊烯
‑2‑
酮;本应用例中,反应转化率为99%;产率为86%,加成产物的光学纯度为81%ee。
[0107]
应用例11
[0108]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为3
‑
庚烯
‑2‑
酮;本应用例中,反应转化率为90%;产率为83%,加成产物的光学纯度为89%ee。
[0109]
应用例12
[0110]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为3
‑
壬烯
‑2‑
酮;本应用例中,反应转化率为81%;产率为73%,加成产物的光学纯度为90%ee。
[0111]
应用例13
[0112]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为查耳酮;本应用例中,反应转化率为81%;产率为76%,加成产物的光学纯度为92%ee。
[0113]
应用例14
[0114]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为1
‑
(4
‑
甲氧基苯基)
‑3‑
苯基丙
‑2‑
烯
‑1‑
酮;本应用例中,反应转化率为78%;产率为73%,加成产物的光学纯度为87%ee。
[0115]
应用例15
[0116]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为4
′‑
氟查耳酮;本应用例中,反应转化率为85%;产率为77%,加成产物的光学纯度为91%ee。
[0117]
应用例16
[0118]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为4
‑
甲氧基查耳酮;本应用例中,反应转化率为94%;产率为90%,加成产物的光学纯度为91%ee。
[0119]
应用例17
[0120]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为4
‑
氯查耳酮;本应用例中,反应转化率为77%;产率为73%,加成产物的光学纯度为91%ee。
[0121]
应用例18
[0122]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为4
‑
甲氧基苯亚甲基丙酮;本应用例中,反应转化率为49%;产率为46%,加成产物的光学纯度为89%ee。
[0123]
应用例19
[0124]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为4
‑
氯苯亚甲基丙酮;本应用例中,反应转化率为61%;产率为54%,加成产物的光学纯度为85%ee。
[0125]
应用例20
[0126]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为1
‑
[3
‑
(三氟甲基)苯基]丁
‑1‑
烯
‑3‑
酮;本应用例中,反应转化率为44%;产率为39%,加成产物的光学纯度为
88%ee。
[0127]
应用例21
[0128]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为2,4
‑
二氯苯亚甲基丙酮;本应用例中,反应转化率为61%;产率为56%,加成产物的光学纯度为95%ee。
[0129]
应用例22
[0130]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为4
‑
(4
‑
羟基
‑3‑
甲氧苯基)
‑3‑
丁烯
‑2‑
酮;本应用例中,反应转化率为65%;产率为58%,加成产物的光学纯度为93%ee。
[0131]
应用例23
[0132]
本应用例与应用例4反应参数相同,区别在于α,β
‑
不饱和酮换为异丁基苯乙烯酮;本应用例中,反应转化率为47%;产率为41%,加成产物的光学纯度为91%ee。
[0133]
对比例1
[0134]
本对比例与与应用例4反应参数相同,区别在于催化剂换为无冠醚衍生的手性1,1
′‑
联
‑
2,2
′‑
萘酚催化剂,结构式如下所示:
[0135][0136]
本对比例中,反应转化率为35%;加成产物的光学纯度为75%ee。
[0137]
以上所述的实施例对本发明的技术方案进行了详细说明,应理解的是以上所述的仅为本发明的具体实施例,并不用于限制本发明,凡在本发明的原则范围内所做的任何修改、补充或类似方式替代等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。