1.本发明涉及医药技术领域,具体为订书肽类抗肿瘤活性化合物及药物组合物与应用,其具有抑制结直肠癌细胞hct116,sw480以及lovo的活性,可用于制备相关抗癌药物。
背景技术:
2.结直肠癌(colorectal cancer,crc)是我国临床上高发的癌症之一,近年来crc的发病和病死的人数逐年增加。在我国,crc发病率在男性和女性中分别排名第4位及第3位,死亡率为第5位及第4位,城镇居民发病率更高。美国统计数据结果显示,在2000年至2013年间,年龄小于50岁的成年人,crc发病率增加了22%,而确诊后5年相对存活率为65%。超过80%结直肠癌发病为散发性的,也与遗传性或炎症性肠病病史相关。结直肠癌的病因涉及饮食习惯、环境因素和慢性炎症性肠病等,其中有腺瘤病史的患者发病风险更高。同时,结直肠癌受遗传因素的影响也很大,在无家族病史的患者中只偶有发生。但目前尚不清楚遗传的具体因素。
3.抗菌肽长期以来一直被认为是治疗人类癌症的很有前途的候选药物,因为它们不仅对多种细菌、真菌、包膜病毒和原生动物具有细胞毒性,而且对不同类型的人类癌细胞也具有细胞毒性。抗菌肽temporin-1cea(fvdlkkianiinsifgk)是从中国褐蛙皮肤分泌物中分离出来的阳离子两亲性α螺旋多肽。前期研究表明,temporin-1cea具有抗革兰氏阴性和阳性细菌活性,以及对mcf-7细胞的抗肿瘤活性。temporin-1cea对人血细胞具有较低的溶血作用和低毒性。然而,线性肽也具有构象不稳定、透膜能力差以及抗水解酶能力弱等问题亟待解决,故而提出订书肽类抗肿瘤活性化合物及其制备方法与应用来解决上述所提出的问题。
技术实现要素:
4.(一)解决的技术问题
5.针对现有技术的不足,本发明提供了订书肽类抗肿瘤活性化合物及药物组合物与应用,具备增强其细胞渗透性、提高酶稳定性和抗肿瘤活性等优点,解决了现有的抗癌多肽药物具有构象不稳定、透膜能力差以及抗水解酶能力弱的问题。
6.(二)技术方案
7.为实现上述增强其细胞渗透性、提高酶稳定性和抗肿瘤活性的目的,本发明提供如下技术方案:订书肽类抗肿瘤活性化合物,具体指具有式(i)结构的订书肽类活性分子及其药学上可接受的盐或酯:
8.fx1x2x3kkx4x5x6x7x8nx9x10fx11k
ꢀꢀ(i)9.其中,x1表示缬氨酸或((2r)-2-氨基-2-甲基-9-癸烯酸;x2表示天冬氨酸或(2r)-2-氨基-2-甲基-6-庚烯酸或(2r)-2-氨基-2-甲基-9-癸烯酸;x3表示亮氨酸或(2r)-2-氨基-2-甲基-6-庚烯酸或(2r)-2-氨基-2-甲基-9-癸烯酸;x4表示异亮氨酸或(2r)-2-氨基-2-甲基-6-庚烯酸或(2r)-2-氨基-2-甲基-9-癸烯酸;x5表示丙氨酸或(2r)-2-氨基-2-甲
基-6-庚烯酸或(2r)-2-氨基-2-甲基-9-癸烯酸;x6表示天冬酰胺或(2r)-2-氨基-2-甲基-6-庚烯酸或(2r)-2-氨基-2-甲基-9-癸烯酸;x7表示异亮氨酸或(2r)-2-氨基-2-甲基-6-庚烯酸;x8表示异亮氨酸或(2r)-2-氨基-2-甲基-6-庚烯酸;x9表示丝氨酸或(2r)-2-氨基-2-甲基-6-庚烯酸;x10表示异亮氨酸或(2r)-2-氨基-2-甲基-6-庚烯酸;x11表示甘氨酸或(2r)-2-氨基-2-甲基-6-庚烯酸;片段中成对的(2r)-2-氨基-2-甲基-6-庚烯酸或(2r)-2-氨基-2-甲基-9-癸烯酸通过烯烃复分解反应进行环合。
10.在本文中,“本发明的订书肽类活性分子”指的是本发明中具有式(i)所示结构多肽,在本文中,这种多肽可以称为“订书肽类活性分子”,“多肽片段”或“本发明多肽。
11.式(i)多肽的n末端的氨基和c末端的羧基以及氨基酸侧链基团可以不进行修饰,也可以在基本不影响本发明多肽活性的前提下进行修饰,如形成“药学上可接受的酯”,n末端氨基基团的修饰包括但不限于脱-氨基、n-低级烷基、n-二低级烷基和n-酰基修饰,c末端羧基基团的修饰包括但不限于酰胺、低级烷基酰胺、二烷基酰胺和低级烷基酯修饰,本发明多肽n末端的氨基进行乙酰化修饰,即是-ac,c末端的羧基进行酰胺化修饰,即是-nh2。
12.本文所使用的多肽及氨基酸和化学基团的表示方法均为所属领域公认的表示方法,其中氨基酸的缩写可参照表1中的定义。特殊氨基酸结构可参照表2中的定义,在本文中,若不特别指出,氨基酸一般指l-型的氨基酸。
13.表1氨基酸缩写表
14.氨基酸三字母缩写一字母缩写氨基酸三字母缩写一字母缩写丙氨酸alaa亮氨酸leul精氨酸argr赖氨酸lysk天冬酰胺asnn蛋氨酸metm天冬氨酸aspd苯丙氨酸phef半胱酰胺cysc脯氨酸prop谷氨酰胺glnq丝氨酸sers谷氨酸glue苏氨酸thrt甘氨酸glyg色氨酸trpw组氨酸hish酪氨酸tyry异亮氨酸ilei缬氨酸valv
15.表2特殊氨基酸缩写表
16.氨基酸缩写(2r)-2-氨基-2-甲基-6-庚烯酸s5(2r)-2-氨基-2-甲基-9-癸烯酸r8[0017]“药物上可接受的盐”指一些小分子酸性或碱性化合物与多肽形成的盐,一般能够增加多肽的溶解性,所形成的盐基本上不改变多肽的活性。
[0018]
例如,通常能与本发明多肽形成盐的酸有盐酸、磷酸、硫酸、乙酸、琥珀酸、马来酸和柠檬酸等;能与本发明多肽形成盐的碱有碱金属或碱土金属的氢氧化物、铵和碳酸盐等。
[0019]
本发明多肽的抗肿瘤作用可以通过所属领域常规的实验方法来验证,如细胞学实验等,在本发明的具体实施方式中,优选通过细胞学实验如cck-8法,通过该试验,发现本发明所涉及的式(i)订书肽类活性分子都具有体外抗肿瘤作用。
[0020]
此外,本发明要解决的另一技术问题是提供了含有式(i)结构的多肽片段的药物组合物,其可以用于抗肿瘤治疗。
[0021]
该组合物可以含有本发明的订书肽类活性分子中的一种或多种,优选仅含一种。
[0022]
该组合物可以含有一种或多种药学上可接受的稀释剂、赋形剂或载体,优选该组合物为单位剂量形式,如片剂、膜剂、丸剂、胶囊(包括持续释放或延迟释放形式)、粉剂、颗粒剂、糖浆剂或乳液剂、消毒的注射用溶液、悬浮液或冻干粉末针剂、气雾剂或液体喷剂、滴剂自动注射装置或栓剂。
[0023]
上述活性药物组分可以与一种无毒的药物学可接受的惰性载体组合在一起,如乙醇、甘油、水或其组合,本发明式(i)的订书肽类活性分子优选使用消毒的注射用水溶液。
[0024]
本发明的药物组合物可通过所属领域技术人员所熟知的给药方式来进行给药,例如口服、直肠、舌下、肺部、透皮、离子透入、阴道及鼻内给药。本发明的药物组合物优选胃肠道外给药,如皮下、肌内或静脉内注射。
[0025]
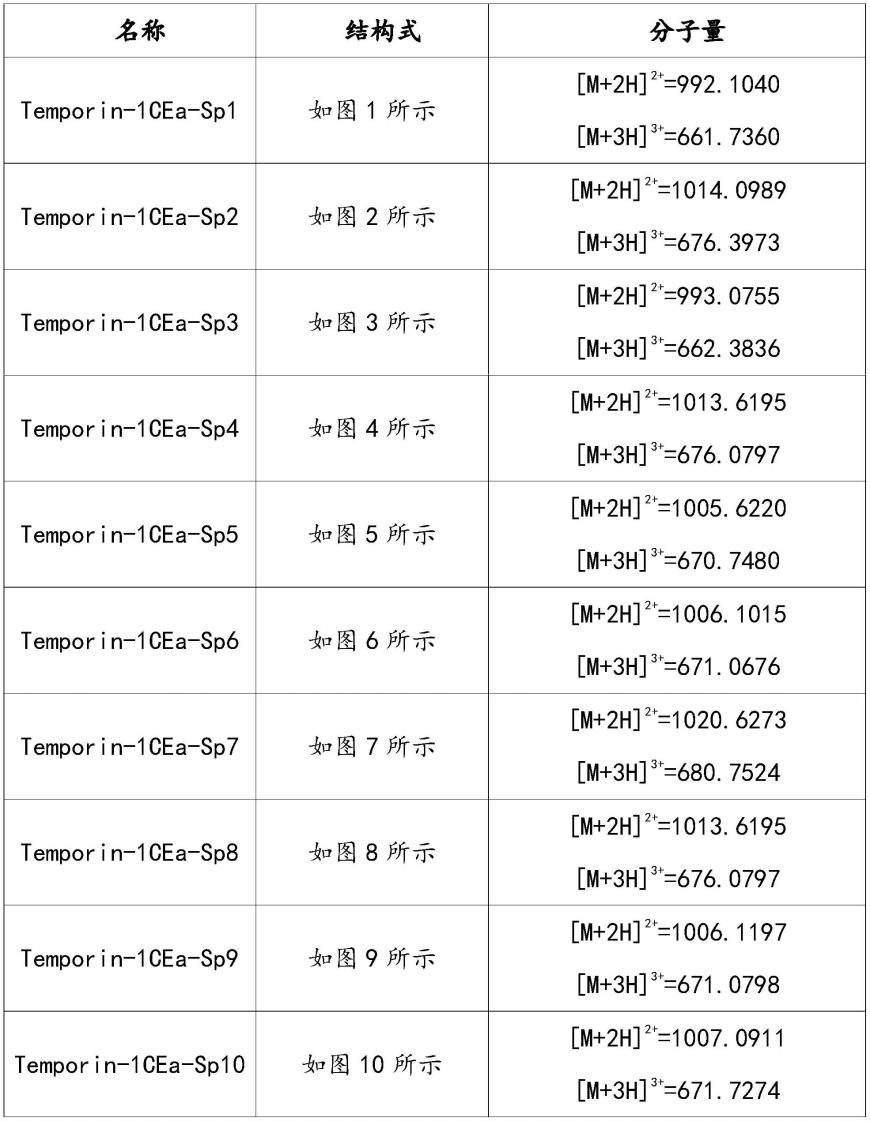
本发明合成的部分优选化合物的名称、结构式和质谱数据如表3所示
[0026]
表3优选订书肽类活性分子的名称、结构式和质谱数据
[0027]
[0028][0029]
为了便于理解,以下将通过具体的实施例和附图对本发明进行描述,需要特别指出的是,这些描述仅仅是示例性的描述,并不构成对本发明范围的限制。
[0030]
(三)有益效果
[0031]
与现有技术相比,本发明提供了订书肽类抗肿瘤活性化合物及其制备方法与应用,具备以下有益效果:
[0032]
该订书肽类抗肿瘤活性化合物及其制备方法与应用,研究报道,通过化学手段增强多肽的α螺旋构型,从而增加其透膜能力和酶稳定性是解决线性多肽成药性差的有效策略,而其中以订书化环合修饰策略报道最多,通过特定氨基酸侧链戊烯基发生烯烃复分解反应进行环合,能够有效提高多肽的结构刚性和巩固α螺旋构型,从而提高酶耐受性和细胞渗透率,因此,我们采用订书化环合修饰策略设计并合成了一系列新型temporin-1cea订书肽类活性分子,旨在增强其细胞渗透性,提高酶稳定性和抗肿瘤活性,更加适用于癌症病人安全用药。
附图说明
[0033]
图1为本发明表3中temporin-1cea-sp1结构式示意图;
[0034]
图2为本发明表3中temporin-1cea-sp2结构式示意图;
[0035]
图3为本发明表3中temporin-1cea-sp3结构式示意图;
[0036]
图4为本发明表3中temporin-1cea-sp4结构式示意图;
[0037]
图5为本发明表3中temporin-1cea-sp5结构式示意图;
[0038]
图6为本发明表3中temporin-1cea-sp6结构式示意图;
[0039]
图7为本发明表3中temporin-1cea-sp7结构式示意图。
[0040]
图8为本发明表3中temporin-1cea-sp8结构式示意图。
[0041]
图9为本发明表3中temporin-1cea-sp9结构式示意图。
[0042]
图10为本发明表3中temporin-1cea-sp10结构式示意图。
[0043]
图11为本发明表3中temporin-1cea-sp11结构式示意图。
[0044]
图12为本发明表3中temporin-1cea-sp12结构式示意图。
[0045]
图13为本发明表3中temporin-1cea-sp13结构式示意图。
具体实施方式
[0046]
下面将结合本发明的实施例,对本发明实施例中的技术方案进行清楚、完整地描
述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0047]
实施例一:订书肽类抗肿瘤活性化合物的制备方法,固相合成temporin-1cea-sp1,具体步骤如下:
[0048]
氨基酸α-氨基用9-芴基甲氧羰基(fmoc)保护,并对氨基酸进行侧链保护:ser的侧链保护基为叔丁基(tbu),lys的侧链保护基为叔丁氧羰基(boc),其中用fmoc-s5-oh替换第三位和第七位的氨基酸,以6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯(hctu)、n,n-二异丙基乙胺(dipea)为活化试剂,使上述保护氨基酸依次偶联,偶联每次40分钟,以20%哌啶/dmf为脱fmoc试剂,每次10分钟,多肽连接完毕后,苯基亚甲基双(三苯己基磷)二氯化钌(第一代grubbs催化剂)作为环合试剂,反应过夜,使用tfa/edt/tips/water(95:2:2:1,v/v/v/v)室温反应2小时,从而将其从树脂上切割下来,同时脱除侧链保护基,后用无水乙醚沉淀得到粗肽,粗肽在30分钟内以反相hplc纯化,冻干得到纯度≥97.0%的白色冻干粉末。
[0049]
实施例二:订书肽类抗肿瘤活性化合物的制备方法,固相合成temporin-1cea-sp8,具体步骤如下:
[0050]
氨基酸α-氨基用9-芴基甲氧羰基(fmoc)保护,并对氨基酸进行侧链保护:ser的侧链保护基为叔丁基(tbu),lys的侧链保护基为叔丁氧羰基(boc),其中用fmoc-r8-oh替换第二位,fmoc-s5-oh替换第九位的氨基酸,以6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯(hctu)、n,n-二异丙基乙胺(dipea)为活化试剂,使上述保护氨基酸依次偶联,偶联每次40分钟,以20%哌啶/dmf为脱fmoc试剂,每次10分钟,多肽连接完毕后,苯基亚甲基双(三苯己基磷)二氯化钌(第一代grubbs催化剂)作为环合试剂,反应过夜,使用tfa/edt/tips/water(95:2:2:1,v/v/v/v)室温反应2小时,从而将其从树脂上切割下来,同时脱除侧链保护基,后用无水乙醚沉淀得到粗肽,粗肽在30分钟内以反相hplc纯化,冻干得到纯度≥97.0%的白色冻干粉末。
[0051]
实验例:
[0052]
1)细胞生物学实验
[0053]
cck-8体外肿瘤抑制实验:结直肠癌细胞hct116,sw480以及lovo分别用含胎牛血清(10%)、青霉素(100ku
·
l-1)及链霉素(100mg
·
l-1)的高糖d-mem培养基于37℃,5%co2培养箱中常规培养传代,取对数生长期的c42b细胞以2
×
104ml-1
的密度接种于96孔板,每孔100μl,每组设3个复孔,多肽以0.39、0.78、1.56、3.125、6.25、12.5、25、50μm浓度作用于细胞,细胞培养96h后,每孔加入含有10%cck-8试剂的完全培养基100μl,37℃,5%co2培养箱避光孵育2h后,用酶标仪(biotek,vermont,usa)在波长450nm处检测各孔吸光度值(od),根据od值计算细胞存活率(vitalrate,vr):vr=(用药组od值-空白组od值)/(对照组od值-空白组od值),计算3个平行孔的平均值vr,根据药物vr,由药物浓度的对数值与vr线性回归求出药物的半数抑制浓度(50%inhibitory concentration,ic
50
)。
[0054]
实验结果:cck-8体外肿瘤抑制实验结果显示,多肽片段均显示出良好的体外肿瘤细胞抑制作用,较阴性对照temporin-1cea均有提高,结果如表4所示。
[0055]
2)酶稳定性实验
[0056]
糜蛋白酶稳定性实验:分别称取1-2mg多肽溶于一定量的dmso中,配置成浓度为
1nm的储存液,称取一定量的糜蛋白酶溶于含有2mm氯化钙的磷酸盐缓冲溶液(50mm,ph=7.4)至糜蛋白的浓度为0.5ng/μl,在2ml离心管中分别加入1950μl含糜蛋白酶的磷酸盐缓冲溶液和50μl的肽储存液进行酶降解反应,分别取0小时、1小时、2小时、4小时、8小时和12小时时间点的50μl反应液加入50μl盐酸(1m)淬灭糜蛋白酶的活性,使用hplc分析不同时间点肽的残余量。
[0057]
实验结果:糜蛋白酶稳定性实验结果显示,temporin-1cea-sp3、temporin-1cea-sp6、temporin-1cea-sp9、temporin-1cea-sp11和temporin-1cea-sp13显示出较阴性对照temporin-1cea更强的抗糜蛋白酶能力,结果如表4所示。
[0058]
表4订书肽类活性分子糜蛋白酶降解半衰期(t
1/2
)以及抗结直肠癌细胞hct116的半数抑制浓度(ic
50
)
[0059][0060][0061]
本发明的有益效果是:该订书肽类抗肿瘤活性化合物及其制备方法与应用,研究报道,通过化学手段增强多肽的α螺旋构型,从而增加其透膜能力和酶稳定性是解决线性多肽成药性差的有效策略,而其中以订书化环合修饰策略报道最多,通过特定氨基酸侧链戊烯基发生烯烃复分解反应进行环合,能够有效提高多肽的结构刚性和巩固α螺旋构型,从而
提高酶耐受性和细胞渗透率,因此,我们采用订书化环合修饰策略设计并合成了一系列新型temporin-1cea订书肽类活性分子,旨在增强其细胞渗透性,提高酶稳定性和抗肿瘤活性,更加适用于癌症病人安全用药,解决了现有的抗癌药物具有构象不稳定、透膜能力差以及抗水解酶能力弱的问题。
[0062]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。