1.本发明涉及生物医学检测领域,具体涉及一种基于脱氧核糖核酶的去甲基化酶活性检测方法和应用。
背景技术:
2.烷基化剂是许多癌症常用的化学药物。当烷基化试剂攻击基因组dna时,烷基化损伤发生在不同的易感位点,如o
6-甲基鸟嘌呤(o6meg)、n
1-甲基腺苷(1mea)和n
3-甲基胞嘧啶(3mec),影响核酸和蛋白质的结构和功能,干扰转录和复制。虽然有一些烷化剂是有效的,但烷化剂耐药性一直是影响肿瘤靶向药物疗效的重要问题。造成这种耐药性的主要因素之一是存在许多内源性去甲基酶,它们修复烷化剂造成的损伤,使癌细胞对烷化剂产生耐药性。其中,o
6-甲基鸟嘌呤dna甲基转移酶(mgmt)和alkb同源蛋白(alkbh)家族被认为是去甲基化蛋白中的两类关键的蛋白,它们在癌症治疗中肿瘤对烷基化药物的化疗耐药性中发挥着重要作用。mgmt是一种dna修复酶,属于转移酶家族。它按1:1化学计量关系将o6meg上的烷基加合物转移到自己的半胱氨酸残基来修复dna中的烷基化鸟嘌呤。研究显示,烷化剂的治疗效果与mgmt表达呈负相关。因此,mgmt活性是评价烷基化药物疗效的重要指标。到目前为止,已经报道了几种检测mgmt活性的方法,包括常用的甲基化特异性pcr(msp)和mgmt活性测试。然而,前者依赖于间接分析mgmt启动子甲基化水平,该甲基化与mgmt活性只有适度的相关性;而后者是一种半定量分析方法,缺乏灵敏性。
3.脱氧核糖核酶(dnazyme)是通过dna文库筛选出来的催化核酸,与蛋白质酶或核酶相似,dnazyme可以催化许多化学和生物转化,如dna或rna切割、连接、磷酸化等,其中一些dnazyme需要特定的辅助因子,如氨基酸、金属离子和有机小分子等,使dnazyme成为开发高选择性生物传感器的新平台。前期研究表明,dnazyme催化核心的碱基序列高度保守,这对维持其催化活性至关重要。在此基础上,对dnazyme进行化学修饰可以调节其催化活性,使其作为一种功能性开关用于开发更多调控型生物化学传感平台。
4.crispr-cas12a(cpf1)可以与单链引导rna(crrna)结合形成cas12a/crrna复合体,不仅像许多其他crispr-cas系统一样切割靶dna,而且被靶dna激活后,cas12a可以切割体系中任何非特异性的单链dna。大多数cas12a系统结合了高效的非特异性ssdna切割技术和核酸扩增技术,检测限低至fm级别,在核酸相关疾病诊断方面具有巨大潜力。
技术实现要素:
5.本发明要解决的技术问题是克服现有技术的不足,提供一种基于脱氧核糖核酶的去甲基化酶活性检测方法和应用。该检测方法基于一种表观遗传修饰敏感dnazyme和crispr/cas12a的信号放大技术,可以实现检测信号的多重放大。同时,该检测方法可实现多种去甲基化酶的的检测以及对烷基化药物的疗效进行评估。
6.为了实现上述目的,本发明提供了一种基于脱氧核糖核酶的去甲基化酶活性检测方法,包括以下步骤:
7.s1、在i-r3 dnazyme催化核心区引入甲基残基获得甲基化的dnazyme;将所述dnazyme与substrate退火杂交,然后与去甲基化酶共孵育过夜反应以去除dnazyme上的甲基,恢复切割活性;
8.s2、加入锌离子形成dnazyme切割反应体系,切割所述substrate的特定位点,释放出可靶向cas12a-crrna的序列;
9.s3、加入含有非特异性单链的cas12a报告溶液,所述可靶向cas12a-crrna的序列靶向激活cas12a,所述cas12a切割所述非特异性单链,使得荧光基团恢复荧光信号,通过监测荧光信号的变化达到检测去甲基化酶活性的目的。
10.上述的锌离子为无水zncl2溶于缓冲溶液后溶液中所含的离子。
11.上述的去甲基化酶活性检测方法,进一步的,所述s1中引入的甲基修饰类型包括o6meg、m6a、1mea、3mec中的一种或多种。
12.上述的去甲基化酶活性检测方法,进一步的,s1中所述去甲基化酶为mgmt、alkbh2、fto中的一种或多种;所述dnazyme为dnazyme1~dnazyme8,dnazyme1.1-4.1中的一种,所述substrate为substrate1~substrate3中的一种;
13.所述dnazyme1的核苷酸序列如seq id no.1所示;
14.所述dnazyme2的核苷酸序列如seq id no.2所示;
15.所述dnazyme3的核苷酸序列如seq id no.3所示;
16.所述dnazyme4的核苷酸序列如seq id no.4所示;
17.所述dnazyme5的核苷酸序列如seq id no.5所示;
18.所述dnazyme1.1的核苷酸序列入seq id no.6所示;
19.所述dnazyme2.1的核苷酸序列入seq id no.7所示;
20.所述dnazyme3.1的核苷酸序列入seq id no.8所示;
21.所述dnazyme4.1的核苷酸序列入seq id no.9所示;
22.所述dnazyme6的核苷酸序列如seq id no.10所示;
23.所述dnazyme7的核苷酸序列如seq id no.11所示;
24.所述dnazyme8的核苷酸序列如seq id no.12所示;
25.所述substrate1的核苷酸序列如seq id no.13所示;
26.所述substrate2的核苷酸序列如seq id no.14所示;
27.所述substrate3的核苷酸序列如seq id no.15所示。
28.进一步的,所述的mgmt为甲基转移酶,可以1:1将dna链上的甲基转移到自身的半胱氨酸残基上。所述的alkbh2、fto为双加氧去甲基化酶,在α-kg,l-抗坏血酸,fe
2
的作用下可以氧化去除dna链上的甲基。
29.上述的去甲基化酶活性检测方法,进一步的,所述s3中所述非特异性单链为携带有荧光基团和荧光淬灭基团的ssdna,所述ssdna的序列如seq id no.17所示,通过检测荧光信号,达到检测去甲基化酶活性的目的。进一步的,所述荧光基团为fam;所述淬灭基团为bhq1。现有技术中,开发出来的荧光基团和淬灭基团种类众多,基本可用于此类实验的标记。
30.上述的去甲基化酶活性检测方法,进一步的,其特征在于,所述cas12a报告溶液包括crrna和cas12a,所述crrna的核苷酸序列如seq id no.16所示。所述的cas12a(又称
cpf1),是crispr/cas体系中一类的具有核酸酶特性的蛋白。
31.上述的去甲基化酶活性检测方法,进一步的,所述cas12a报告溶液的成分还包括:50mm hepes、100mm nacl、20mm mgcl2,ph 7.4。
32.上述的去甲基化酶活性检测方法,进一步的,所述s1具体为:
33.s1-1、将dnazyme与substrate混合于去甲基化反应的缓冲体系中,置于95℃反应5min。
34.s1-2、加入去甲基化酶,37℃过夜反应。
35.上述的去甲基化酶活性检测方法,进一步的,所述s1-2可替换为:加入细胞裂解液,37℃过夜反应。
36.上述的去甲基化酶活性检测方法,进一步的,所述细胞裂解液采用以下方法制备得到:使用胰酶-edta分别收集t98g,u-87,mcf-7,293t细胞悬液,在1500rpm,4℃下离心5min,沉淀中加入passive lysis buffer和蛋白酶抑制剂pmsf,置于冰上裂解30min,后在15000rpm,4℃下离心15min,收集上清液即为含有细胞总蛋白的细胞裂解液。使用bca蛋白定量试剂盒对细胞裂解液中的总蛋白进行定量。所选用的细胞系包括t98g,u-87,mcf-7,293t细胞系。t98g和u-87都属于胶质瘤细胞系,已知mgmt蛋白在t98g细胞系中高表达,在u-87细胞系中低表达;mcf-7属于人乳腺癌细胞系,也是高表达mgmt的细胞系;293t属于人肾上皮细胞系,是低表达mgmt的细胞系。
37.上述的去甲基化酶活性检测方法,进一步的,mgmt去甲基化反应的缓冲体系的成分包括:50mm hepes和100mm nacl,ph 7.0。
38.alkbh2去甲基化反应的缓冲体系的成分包括:50mm hepes、100mm nacl、2mm ascorbate、1mmα-kg、75μm(nh4)2fe(so4)2、0.1mg/ml bsa,ph 8.0。
39.上述的去甲基化酶活性检测方法,进一步的,所述dnazyme切割反应体系的成分包括:50mm hepes、100mm nacl、2mm zncl2,ph 7.0。
40.上述的去甲基化酶活性检测方法,进一步的,所述锌离子的浓度为2mm~10mm。进一步的,所述锌离子浓度为2mm。
41.上述的去甲基化酶活性检测方法,进一步的,所述substrate:dnazyme浓度比为1:1~20:1。进一步的,所述substrate:dnazyme浓度比为5:1。
42.基于一个总的技术构思,本发明还提供了一种上述去甲基化酶活性检测方法在对烷基化药物的疗效进行评估中的应用。
43.与现有技术相比,本发明的优点在于:
44.(1)本发明提供了一种基于脱氧核糖核酶的去甲基化酶活性检测方法,相较于现有技术中对去甲基化酶的半定量检测和去甲基化酶启动子甲基化检测,本发明利用去甲基化酶可去除dna链上甲基残基的特性,将甲基化修饰的dnazyme设计成一个活性可控的开关,而去甲基化酶则作为开启整个信号响应通道的钥匙,实现对去甲基化酶的定量检测。
45.(2)本发明提供了一种基于脱氧核糖核酶的去甲基化酶检测方法,利用了dnazyme核酸酶的特性,实现信号的一重放大。
46.(3)本发明提供了一种基于脱氧核糖核酶的去甲基化酶检测方法,结合了crispr/cas12a的高效反式切割信号扩增技术,通过dnazyme切割释放出的靶向cas12a的序列激活cas12a,随后,cas12a对带有荧光基团和淬灭基团的ssdna(fam-ssdna-bhq1)进行切割,实
现信号二重放大,提高了检测的灵敏度。
47.(4)本发明提供了一种基于脱氧核糖核酶的去甲基化酶检测方法,通过设计不同类型的甲基修饰,可以对不同的去甲基化酶进行检测。
48.(5)本发明提供了一种基于脱氧核糖核酶的去甲基化酶检测方法,不仅可在缓冲溶液中进行mgmt活性检测,还可以在复杂的体系,例如细胞裂解液中检测mgmt活性,为研究细胞内mgmt活性提供了新的检测方法。
49.综上,本发明的基于脱氧核糖核酶的去甲基化酶活性检测方法具有操作简单,检测成本低、检测灵敏度高等优点,可作为去甲基化酶检测的一种范式。
附图说明
50.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整的描述。
51.图1为本发明实施例2的原理图。
52.图2为本发明实验一中i-r3 dnazyme结构示意图、本发明所选择的o6g甲基残基插入位点示意图和筛选活性抑制位点的凝胶图。
53.图3为本发明实验二中mgmt介导dnazyme活性恢复的凝胶图和dnazyme活性动力学表征图。
54.图4为本发明实验三中i-r3 dnazyme锌离子依赖的荧光响应图。
55.图5为本发明实验四中mgmt介导dnazyme1.1-3.1活性恢复的荧光恢复图和substrate和dnazyme2.1(o6meg16)的比例优化图。
56.图6为本发明实验五中基于dnazyme-crispr/cas12a系统检测不同浓度的mgmt的荧光图和基于dnazyme-crispr/cas12a系统检测mgmt的标准曲线图。
57.图7为本发明实验六中基于dnazyme-crispr/cas12a系统检测mgmt的选择性响应荧光图。
58.图8为本发明实验七中mgmt选择性修复o6meg甲基化的荧光图。
59.图9为本发明实验八中在细胞裂解液中mgmt蛋白活性检测的标准曲线;检测不同数量的t98g细胞裂解液中mgmt活性的柱状图;检测不同细胞系中mgmt活性的柱状图;dnazyme-crispr/cas12a系统检测药物处理前后细胞内mgmt活性变化的柱状图。
60.图10为本发明实施例5的原理图。
61.图11为本发明实验九中所选择的1mea,3mec甲基残基插入位点示意图和筛选活性抑制位点的凝胶图。
62.图12为本发明实验十中alkbh2介导的dnazyme7(1mea17)活性恢复的凝胶图和alkbh2介导dnazyme7(1mea17)活性恢复的荧光恢复图。
具体实施方式
63.以下结合具体优选的实施例对本发明作进一步描述,但并不因此而限制本发明的保护范围。
64.除非另有定义,下文中所使用的所有专业术语与本领域技术人员通常理解含义相同。本文中所使用的专业术语只是为了描述具体实施例的目的,并不是旨在限制本发明的
保护范围。
65.除非另有特别说明,本发明中用到的各种原材料、试剂、仪器和设备等均可通过市场购买得到或者可通过现有方法制备得到。下述实施例中的方法,如无特别说明,均为本领域的常规方法。
66.其中fs5荧光仪购于edinburgh公司;l-抗坏血酸、(nh4)2fe(so4)2·
6h2o、α-kg、无dna/rna酶水、6
×
loading buffer、10
×
tbe粉末购于生工生物工程有限公司。
67.本文所用到dna和rna序列均在北京擎科生物科技有限公司、安徽通用生物有限公司、湖南艾科瑞生物有限公司合成并采用page或hplc法纯化。mgmt蛋白购于cayman chemical;alkbh2,fto购于active motif;牛血清蛋白(bsa)购于new england biolabs;其他的化学试剂购于生工生物有限公司。
68.dulbecco's modified eagle培养基(dmem),rpmi 1640培养基,胎牛血清(fbs)和青霉素,链霉素购于invitrogen(carlsbad,usa);胰酶购于genview(usa);dexamethasone(dxm)购于sigma aldrich chemical co.(st louis,usa);passive lysis buffer,pmsf,bca蛋白定量试剂盒购于上海碧云天生物技术有限公司。
69.mcf-7细胞购于national collection of authenticated cell cultures with str analysis(shanghai,china).t98g,u-87,293t细胞购于procell life science&technology co.,ltd(wuhan,china)。
70.实施例1:
71.用于筛选甲基活性抑制位点的脱氧核糖核酶dnazyme1-4:
72.dnazyme1(for page)caagataatctagttgagctgtctgca(seq id no.1)。
73.dnazyme2(o6meg13)caagataatctag
me
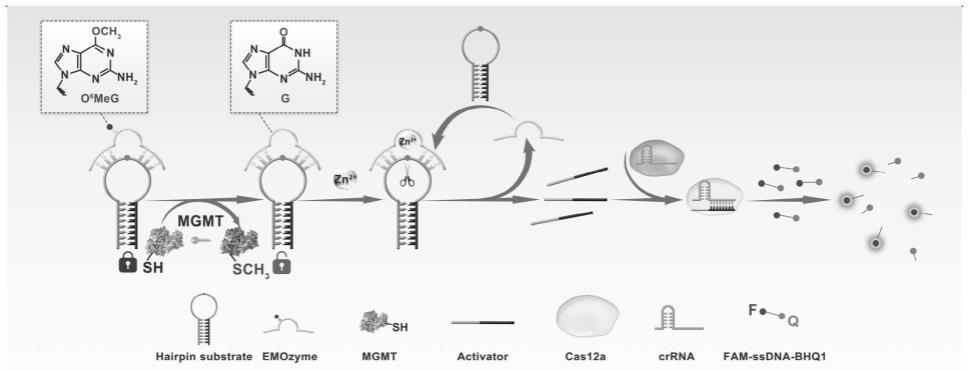
ttgagctgtctgca(seq id no.2)。
74.dnazyme3(o6meg16)caagataatctagttg
me
agctgtctgca(seq id no.3)。
75.dnazyme4(o6meg18)caagataatctagttgag
me
ctgtctgca(seq id no.4)。
76.划线部分是dnazyme的催化核心区,me位置为上标前一个碱基。
77.实施例2:
78.一种基于脱氧核糖核酶的检测方法用于mgmt蛋白的检测,其检测原理参见图1,检测方法包括以下步骤:
79.(1)mgmt去甲基化反应:
80.1.1、将5μm substrate和1μm dnazyme混于50mm hepes,100mm nacl,ph 7.00缓冲溶液中,在95℃反应5min后自然冷却至室温。
81.1.2、于上述溶液中加入1μm的mgmt,于37℃下过夜反应。
82.(2)dnazyme的切割反应:
83.于上述反应中加入锌离子,使得反应体系为:2mm zn
2
,50mm hepes,100mm nacl,ph 7.0,在37℃下反应3h。
84.(3)crispr/cas12a的反式切割反应:
85.3.1、配制crispr/cas12a的报告溶液:
86.将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,形成cas12a报告溶液。
87.crrna:uaa uuu cua cua agu gua gauuca acuu gug ugu uua ccu ggg(seq id no.16)。
88.fam-ssdna-bhq1:fam-ttatt-bhq1(seq id no.17)。
89.3.2、将上述的cas12a报告溶液加入至步骤(2)反应完成后的体系中,在25℃下反应1.5h。
90.(4)荧光检测:
91.于上述反应中加入去离子水至总体积为100μl,随后于490nm波长下激发,收集505nm-660nm处的荧光光谱。
92.图1为本发明实施例2的原理图:通过在i-r3 dnazyme催化核心区引入甲基残基,通过聚丙烯酰胺凝胶电泳(page)表征i-r3 dnazyme对底物链的切割效率,以此筛选出能够抑制i-r3 dnazyme切割活性的甲基位点。筛选出来的dnazyme活性抑制位点被引入o6g甲基残基,通过与去甲基化酶mgmt共孵育可去除dnazyme上的甲基残基以恢复切割活性。dnazyme活性恢复后,在锌离子的存在下可切割底物链,释放出可靶向cas12a-crrna的序列以激活cas12a的核酸酶活性。激活的crispr/cas12a体系可对体系中存在的非特异性单链fam-ssdna-bhq1进行切割,使得荧光基团和淬灭基团分离,从而恢复荧光基团的荧光,获得荧光信号。
93.实验一:筛选甲基敏感的dnazyme的活性抑制位点。通过聚丙烯酰胺凝胶(page)电泳进行表征,具体方法为:
94.(1)将1μm substrate1分别和1μm dnazyme1~4混于50mm hepes,100mm nacl,ph 7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
95.substrate1(for page)的核苷酸序列如seq id no.13所示,具体为:
96.cttcttccctaaccctaaccctaacccttttttttttttttttttttttttttttttcatctcttgcagacgttgaaggattatcttg。
97.(2)dnazyme的切割反应:于上述反应体系中加入锌离子,使得反应体系为2mm zn
2
,50mm hepes,100mm nacl,ph 7.0,在37℃下反应3h。
98.(3)page表征:
99.将上述反应所得溶液用12%page在150v下进行45min,用凝胶成像仪进行成像。
100.图2(a)为i-r3 dnazyme结构示意图和本实验所选择的o6g甲基残基插入位点示意图;(b)为本实验筛选活性抑制位点的凝胶图,从图2b可以看出,筛选出来的三个位点的甲基化(o6meg13、o6meg16、o6meg18)都没有产生底物切割片断,说明这三个位点都能有效抑制dnazyme的活性。
101.实验二:验证mgmt是否能够有效去除dnazyme上的甲基残基,恢复其活性。
102.1.page表征验证mgmt去除dnazyme上的甲基以恢复其活性:
103.(1)将1μm substrate1分别和1μm dnazyme1~4混于50mm hepes,100mm nacl,ph 7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
104.(2)上述dnazyme2~4的反应溶液中加入10μm mgmt,于37℃下过夜反应,且dnazyme2~4体系各设置一个不加mgmt的对照组。
105.(3)于上述反应中加入锌离子,使得反应体系为2mm zn
2
,50mm hepes,100mm nacl,ph 7.0,在37℃下反应3h。
106.(4)page表征:
107.将上述反应所得溶液用12%page在150v下进行45min,用凝胶成像仪进行成像。
108.2.dnazyme切割活性动力学表征。
109.(1)将1μm substrate1分别和1μm dnazyme1~4于50mm hepes,100mm nacl,ph7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
110.(2)往上述dnazyme2~4的溶液中加入10μm mgmt,于37℃下过夜反应,且dnazyme2~4体系各设置一个不加mgmt的对照组。
111.(3)于上述反应体系中加入锌离子,使得反应体系为2mm zn
2
,50mm hepes,100mm nacl,ph 7.0,在37℃下反应3h。
112.(4)page表征:
113.将上述反应所得溶液用12%page在150v下进行45min,用凝胶成像仪进行成像。
114.图3中(a)为本发明mgmt介导dnazyme活性恢复的凝胶图,从图中可以看出,筛选出来的三个位点(g13、g16、g18)都能通过mgmt去除甲基化而恢复dnazyme的活性;(b)为本发明dnazyme活性动力学表征图,从图中可以看出,三个引入甲基残基的dnazyme(g13、g16、g18)活性被完全抑制,而通过mgmt修复后,其活性恢复。
115.实验三:dnazyme是一种锌离子依赖的水解酶,验证dnazyme是否只有在锌离子的存在下才能切割底物。
116.(1)将5μm substrate2和1μm dnazyme5混于50mm hepes,100mm nacl,ph 7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
117.dnazyme5的核苷酸序列如seq id no.5所示,具体为:
118.cgtcatgataatctagttgagctgtcagcagtctgca。
119.substrate2(for fluoresence)的核苷酸序列如seq id no.14所示,具体为:
120.cccaggtaaacacacaagttgattttttttgcagactgctgacgttgaaggattatc atgacgtttttttaacttgtgtgtttacctggg。
121.(2)于上述反应中加入不同锌离子浓度(0μm,50μm,100μm,1mm,2mm,5mm,10mm)的缓冲溶液,在37℃下反应3h。
122.(3)crispr/cas12a的反式切割反应:将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph 7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,得到cas12a报告溶液。
123.(4)将上述的cas12a报告溶液加入至步骤(2)反应完成后的体系中,在25℃下反应1.5h,通过fs5荧光光谱仪检测荧光。
124.图4为i-r3 dnazyme锌离子依赖的荧光响应图。从图中可以看出,在无锌离子存在下,无荧光信号恢复,随着锌离子浓度增加,荧光信号依赖性增加,且在锌离子浓度为2mm时,荧光信号最强,因此选择2mm锌离子作为本实验最佳离子浓度。
125.实施例3:
126.一种用于荧光检测mgmt蛋白活性的脱氧核糖核酶dnazyme5、dnazyme1.1~3.1:
127.dnazyme5:cgtcatgataatctagttgagctgtcagcagtctgca(seq id no.5)。
128.dnazyme1.1:cgtcatgataatctag
me
ttgagctgtcagcagtctgca(seq id no.6)。
129.dnazyme2.1:cgtcatgataatctagttg
me
agctgtcagcagtctgca(seq id no.7)。
130.dnazyme3.1:cgtcatgataatctagttgag
me
ctgtcagcagtctgca(seq id no.8)。
131.由实验二可知,mgmt能恢复g13,g16,g18位点o6g甲基化修饰的dnazyme的活性。为了验证本发明可以通过荧光信号检测mgmt活性,设计了以上序列用于荧光检测实验。dnazyme5为未引入甲基残基的阳性对照,dnazyme1.1-3.1为在g13,g16,g18位点引入甲基的实验组,划线部分为dnazyme链的催化核心序列。其检测方法与实施例2一致。
132.实验四、考察mgmt介导的dnazyme1.1~3.1的活性恢复及条件优化。
133.1.验证mgmt是否能够恢复dnazyme1.1~3.1的活性。
134.(1)将5μm substrate2分别和1μm dnazyme1.1~3.1,dnazyme5混于50mm hepes,100mm nacl,ph 7.0缓冲溶液中,在95℃反应5min后自然冷却至室温。
135.(2)上述dnazyme1.1-3.1的反应溶液中加1μm mgmt,于37℃下过夜反应,同时各设置一组不加mgmt的对照组。
136.(3)于上述反应中加入锌离子,使得反应体系为2mm zn
2
,50mm hepes,100mm nacl,ph 7.00,在37℃下反应3h。
137.(4)crispr/cas12a的反式切割反应:
138.将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,获得cas12a报告溶液。
139.(5)将上述的cas12a报告溶液加入至步骤(3)反应完成后的体系中,在25℃下反应1.5h,通过fs5荧光光谱仪检测荧光。
140.2.优化substrate和dnazyme2.1的比例,提高信背比。
141.(1)将1μm dnazyme2.1与不同浓度的substrate2(1μm,5μm,10μm,20μm)混于50mm hepes,100mm nacl,ph 7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
142.(2)上述溶液中加1μm mgmt,于37℃下过夜反应。
143.(3)于上述反应中加入锌离子,使得反应体系为2mm zn
2
,50mm hepes,100mm nacl,ph 7.0,在37℃下反应3h。
144.(4)crispr/cas12a的反式切割反应:
145.将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,获得cas12a报告溶液。
146.(5)将上述的cas12a报告溶液加入至步骤(3)反应完成后的体系中,在25℃下反应1.5h,通过fs5荧光光谱仪检测荧光。
147.图5中(a)为mgmt介导dnazyme1.1-3.1活性恢复的荧光恢复图。图中wild type组为dnazyme5在有无锌离子存在下的荧光柱状对比图,作为阳性对照。从图中可看出,在mgmt存在下,三个不同位置甲基化的dnazyme体系都产生了明显的信号增强,并且dnazyme2.1的荧光信背比最高,所以选择dnazyme2.1用于后续的实验。(b)为本发明的substrate和dnazyme2.1的比例优化图,由图可知,在substrate:dnazyme为5:1时,产生的荧光信背比最高,所以后续实验都采用此最佳比例。
148.实验五:mgmt的灵敏度测试。
149.(1)将5μm substrate2和1μm dnazym2.1(o6meg16)混于50mm hepes,100mm nacl,
no.12)。
168.(2)上述溶液中加入1μm mgmt,于37℃下过夜反应。
169.(3)于上述反应中加入锌离子,使得反应体系为2mm zn
2
,50mm hepes,100mm nacl,ph 7.0,在37℃下反应3h。
170.(4)crispr/cas12a的反式切割反应:将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph 7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,获得报告溶液。
171.(5)将上述的cas12a报告溶液加入至步骤(3)反应完成后的体系中,在25℃下反应1.5h,通过fs5荧光光谱仪检测荧光。
172.图8为本发明的mgmt选择性修复o6meg甲基化的荧光图。从图中可以看出:mgmt可以选择性去除o6meg甲基化,对m6a、1mea、3mec没有选择性,说明本发明开发的体系的选择性高。
173.实验八、细胞裂解液中mgmt的活性检测。
174.(1)制作细胞裂解液中的标准曲线。
175.1.1、细胞培养:分别将t98g细胞、u-87细胞、293t细胞用deme培养基加10%胎牛血清(fbs)和100u/ml青霉素、链霉素(penicillin and streptomycin)培养。mcf-7细胞用rpmi 1640培养基加10%胎牛血清(fbs)和100u/ml青霉素、链霉素培养。细胞培养环境维持在37℃,5%co2。
176.1.2、细胞裂解液中总蛋白提取:将培养后的t98g细胞、u-87细胞、293t细胞、mcf-7细胞通过胰酶-edta消化后在1500rpm下离心5min,舍弃上清液,加入passive lysis buffer和pmsf冰上裂解30min,然后在4℃,15000rpm下离心15min。所得上清液即为含有细胞总蛋白的细胞裂解液。通过bca蛋白定量试剂盒对上述所得总蛋白进行定量。
177.1.3、为制作标准曲线,本实验选取了已知的mgmt低表达的细胞系u-87(胶质瘤细胞系),按上述总蛋白提取步骤,取大约5
×
105个u-87细胞提取总蛋白,得到含100μg总蛋白的细胞裂解液。
178.1.4、将5μm substrate2和1μm dnazyme2.1(o6meg16)混于50mm hepes,100mm nacl,ph 7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
179.1.5、上述溶液中加入到步骤(1.3)的细胞裂解液中,分别加入不同浓度的mgmt(0nm、1nm、5nm、10nm、20nm、50nm),于37℃下过夜反应。
180.1.6、于上述反应体系中加入2mm zn
2
,在37℃下反应3h。
181.1.7、crispr/cas12a的反式切割反应:
182.将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,获得cas12a报告溶液。
183.1.8、将上述的cas12a报告溶液加入至步骤(1.6)反应完成后的体系中,在25℃下反应
184.1.5h,通过fs5荧光光谱仪检测荧光。
185.(2)检测不同细胞数所得裂解液中的mgmt活性。
186.2.1、本实验选取已知的mgmt蛋白高表达的细胞系t98g(胶质瘤细胞系),分别取5
×
105、1
×
106、5
×
106个t98g细胞制备细胞裂解液。
187.2.2、将5μm substrate2和1μm dnazyme2.1(o6meg16)混于50mm hepes,100mm nacl,ph 7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
188.2.3、上述溶液中分别加入到步骤(2.1)的细胞裂解液中,于37℃下过夜反应。
189.2.4、于上述反应体系中加入2mm zn
2
,在37℃下反应3h。
190.2.5、crispr/cas12a的反式切割反应:
191.将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,获得cas12a报告溶液。
192.2.6、将上述的cas12a报告溶液加入至步骤(2.4)反应完成后的体系中,在25℃下反应1.5h,通过fs5荧光光谱仪检测荧光。
193.(3)检测不同细胞系裂解液中mgmt的活性。
194.3.1、分别取约5
×
105个t98g细胞、u-87细胞、293t细胞、mcf-7细胞制备细胞裂解液。
195.3.2、将5μm substrate2和1μm dnazyme2.1(o6meg16)混于50mm hepes,100mm nacl,ph 7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
196.3.3、上述溶液中分别加入到步骤(3.1)的细胞裂解液中,于37℃下过夜反应。
197.3.4、于上述反应体系中加入2mm zn
2
,在37℃下反应3h。
198.3.5、crispr/cas12a的反式切割反应:
199.将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,获得cas12a报告溶液。
200.3.6、将上述的cas12a报告溶液加入至步骤(3.4)反应完成后的体系中,在25℃下反应1.5h,通过fs5荧光光谱仪检测荧光。
201.(4)探究本发明开发的dnazyme-crispr/cas12a系统评估药物处理后的细胞中mgmt活性变化的能力。
202.4.1、选择已知的治疗胶质瘤的药物dxm,该药物是一种烷基化药物,可以使dna产生不同程度的烷化病变,从而杀死细胞达到治疗癌症的作用。而mgmt活性则被认为是导致胶质瘤治疗药物抗性的主要因素,高的mgmt修复活性导致胶质瘤细胞对烷基化药物有更高的耐受性,使得药物疗效降低,通过监控细胞内mgmt活性有助于评估烷基化药物治疗的疗效。本实验选择状态良好的u-87细胞分别与不同的浓度的dxm(0μm,0.1μm,0.5μm,1μm)共孵育24h,然后取约5
×
105个细胞裂解制备细胞裂解液。
203.4.2、将5μm substrate2和1μm dnazyme2.1(o6meg16)混于50mm hepes,100mm nacl,ph 7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
204.4.3、上述溶液中分别加入到步骤(4.1)的细胞裂解液中,于37℃下过夜反应。
205.4.4、于上述反应体系中加入2mm zn
2
,在37℃下反应3h。
206.4.5、crispr/cas12a的反式切割反应:
207.将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,获得cas12a报
告溶液。
208.4.6、将上述的cas12a报告溶液加入至步骤(4.4)反应完成后的体系中,在25℃下反应1.5h,通过fs5荧光光谱仪检测荧光。
209.图9中(a)为本发明在细胞裂解液中mgmt蛋白活性检测的标准曲线,根据此标准曲线的线性拟合公式y=0.1297*x 1.581,可以通过荧光信号倍数计算出细胞裂解液中mgmt活性(通过浓度表示)。(b)为本发明的检测不同数量的t98g细胞裂解液中mgmt活性的柱状图。由图可知,mgmt活性与细胞数量呈正相关。(c)为本发明的检测不同细胞系中mgmt活性的柱状图。由图可知,mgmt分别在t98g和u-87细胞系中高表达和低表达;mgmt在mcf-7细胞系的表达也高于293t细胞系,这与之前的报道一致,说明本发明的基于dnazyme-crispr/cas12a的检测系统能很好的检测不同细胞系中mgmt的活性。(d)为本发明的dnazyme-crispr/cas12a系统检测药物处理前后细胞中mgmt活性变化的柱状图。由图可知,在细胞适应范围内,dxm刺激了mgmt表达的上调,从而提高了其去甲基化活性水平,这与之前的报道一致,说明本发明开发的基于dnazyme-crispr/cas12a的检测系统能够检测细胞内mgmt活性的变化,为胶质瘤的诊断治疗提供了一个灵敏的检测方式。
210.实施例4:
211.一种用于alkbh2蛋白检测的脱氧核糖核酶dnazyme6~dnazyme8:
212.dnazyme6(1mea12):cgtcatgataatcta
me
gttgagctgtcagcagtctgca(seq id no.10)。
213.dnazyme7(1mea17):cgtcatgataatctagttga
me
gctgtcagcagtctgca(seq id no.11)。
214.dnazyme8(3mec19):cgtcatgataatctagttgagc
me
tgtcagcagtctgca(seq id no.12)。
215.实施例5:
216.一种本发明的基于脱氧核糖核酶的检测方法用于alkbh2蛋白的检测,检测原理参见图10,其检测方法包括以下步骤:
217.(1)alkbh2去甲基化反应:
218.1.1、将5μm substrate和1μm dnazyme混于50mm hepes,100mm nacl,ph 7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
219.1.2、于上述溶液中加入2μm alkbh2,2mm l-抗坏血酸,1mmα-kg,75μm(nh4)2fe(so4)2,0.1mg/ml bsa于37℃下过夜反应。
220.(2)dnazyme的切割反应:
221.于上述反应中加入锌离子,使得反应体系为2mm zn
2
,50mm hepes,100mm nacl,ph7.0,在37℃下反应3h。
222.(3)crispr/cas12a的反式切割反应:
223.3.1、配制crispr/cas12a的报告溶液:
224.将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,形成cas12a报告溶液。
225.3.2、将上述的cas12a报告溶液加入至步骤(2)反应完成后的体系中,在25℃下反
应1.5h。
226.(4)荧光检测:于上述反应中加入去离子水至总体积为100μl,随后于490nm波长下激发,收集505nm-660nm处的荧光光谱。
227.图10为本发明的实施例5的原理图。通过在i-r3 dnazyme催化核心区引入甲基残基,通过聚丙烯酰胺凝胶电泳(page)表征i-r3 dnazyme对底物链的切割效率,以此筛选出能够抑制i-r3 dnazyme切割活性的甲基位点。筛选出来的dnazyme活性抑制位点被引入1mea或3mec甲基残基,通过与去甲基化酶alkbh2共孵育可去除dnazyme上的甲基残基以恢复切割活性。dnazyme活性恢复后,在锌离子的存在下可切割底物链,释放出可靶向cas12a-crrna的序列以激活cas12a的核酸酶活性。激活的crispr/cas12a体系可对体系中存在的非特异性单链fam-ssdna-bhq1进行切割,使得荧光基团和淬灭基团分离,从而恢复荧光基团的荧光,获得荧光信号。
228.实验九:筛选甲基敏感的dnazyme活性抑制位点,通过聚丙烯酰胺凝胶(page)电泳进行表征。
229.(1)将1μm substrate3和1μm dnazyme6~dnazyme 8混于50mm hepes,100mm nacl,ph 7.0的缓冲溶液中,在95℃反应5min后自然冷却至室温。
230.substrate3(1mea/3mec)的核苷酸序列如seq id no.15所示,具体为:
231.ttgattttttttgcagactgctgacgttgaaggattatcatgacgtttttttaact。
232.(2)dnazyme的切割反应:
233.于上述反应中加入锌离子,使得反应体系为2mm zn
2
,50mm hepes,100mm nacl,ph7.0,在37℃下反应3h。
234.(3)page表征:将上述反应所得溶液用12%page在150v下进行45min,用凝胶成像仪进行成像。
235.图11中(a)为本发明所选择的1mea,3mec甲基残基插入位点示意图;(b)为本发明的筛选活性抑制位点的凝胶图,从图中可以看出,筛选出来的三个位点的甲基化(1mea12,1mea17,3mec19)中,只有1mea17能有效抑制i-r3 dnazyme的活性。
236.实验十:验证alkbh2是否能够有效去除dnazyme7(1mea17)上的甲基,恢复其活性。
237.(1)通过page表征验证alkbh2恢复dnazyme7(1mea17)的活性。
238.1.1、将1μm substrate3和1μm dnazyme7,dnazyme5混于50mm hepes,100mm nacl缓冲溶液中,在95℃反应5min后自然冷却至室温。
239.1.2、于上述dnazyme7的反应溶液中加入含2μm alkbh2,2mm l-抗坏血酸,1mmα-kg,75μm(nh4)2fe(so4)2,0.1mg/ml bsa的缓冲溶液(50mm hepes,100mm nacl ph8.0),在37℃下过夜反应,同时设置一组不加alkbh2的对照组。
240.1.3、于上述反应中加入锌离子,使得反应浓度为2mm zn
2
,50mm hepes,100mm nacl,ph 7.0,在37℃下反应3h。
241.1.4、page表征:将上述反应所得溶液用12%page在150v下进行45min,用凝胶成像仪进行成像。
242.(2)通过荧光检测验证alkbh2恢复dnazyme7(1mea17)的活性:
243.2.1、将5μm substrate2和1μm dnazyme7混于50mm hepes,100mm nacl缓冲溶液中,在95℃反应5min后自然冷却至室温。
244.2.2、上述反应中加入含2μm alkbh2,2mm l-抗坏血酸,1mmα-kg,75μm(nh4)2fe(so4)2,0.1mg/ml bsa的缓冲溶液(50mm hepes,100mm nacl ph 8.0),在37℃下过夜反应。
245.2.3、于上述反应中加入锌离子,使得反应浓度为2mm zn
2
,50mm hepes,100mm nacl,ph 7.0,在37℃下反应3h。
246.2.4、crispr/cas12a的反式切割反应:
247.将200nm cas12a与400nm crrna混于50mm hepes,100mm nacl,20mm mgcl2(ph7.4)的缓冲溶液中,在25℃下反应30min,然后加入2μm fam-ssdna-bhq1,形成cas12a报告溶液。
248.将上述的cas12a报告溶液加入至步骤(2.3)反应完成后的体系中,在25℃下反应1.5h,通过fs5荧光光谱仪检测荧光。
249.图12中(a)为本发明为alkbh2介导的dnazyme7(1mea
17
)活性恢复的凝胶图。由图可知,在alkbh2存在下,dnazyme7能够切割底物产生切割片段,说明alkbh2能够恢复dnazyme7的活性。(b)为本发明的alkbh2介导dnazyme7(1mea
17
)活性恢复的荧光恢复图。由图可知,alkbh2能有效去除dnazyme7(1mea
17
)上的甲基残基,从而介导了荧光信号的恢复。
250.以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制。虽然本发明已以较佳实施例揭示如上,然而并非用以限定本发明。任何熟悉本领域的技术人员,在不脱离本发明的精神实质和技术方案的情况下,都可利用上述揭示的方法和技术内容对本发明技术方案做出许多可能的变动和修饰,或修改为等同变化的等效实施例。因此,凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所做的任何简单修改、等同替换、等效变化及修饰,均仍属于本发明技术方案保护的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。