1.本发明属于有机合成技术领域,具体涉及一种无溴盐酸多巴胺合成新方法,其以2-苯甲氧基苯酚为原料,经氯甲基化、氰化、还原并与盐酸成盐得盐酸多巴胺。与传统方法相比,该方法具有合成路线短、无溴和收率高的特点,适合工业生产。
背景技术:
2.多巴胺是内源性中枢神经性递质,具有兴奋β-受体、α-受体和多巴胺受体的作用。兴奋心脏β-受体可增加心肌收缩力,增加心输出量。兴奋多巴胺受体和α-受体使肾、肠系膜、冠状动脉及脑血管扩张、血流量增加。对周围血管有轻度收缩作用,升高动脉血压。本药的突出作用为增加肾血流量,肾小球滤过率增加,从而促使尿量增加,尿钠排泄也增加。临床用于各种类型的休克,尤其适用于休克伴有心收缩力减弱,肾功能不全者,临床上用其盐酸盐,即盐酸多巴胺1,结构式如下所示:。
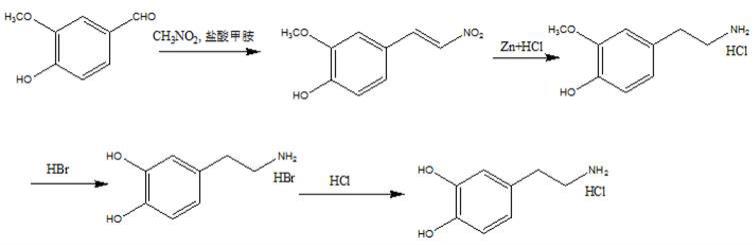
3.盐酸多巴胺现用化学合成,其路线主要由以下几种:路线一(全国原料药工艺汇编,国家医药管理总局,1980年,497~499):以香兰素(3-甲氧基-4-羟基苯甲醛)为原料,在盐酸甲胺催化下与硝基甲烷缩合,然后在盐酸中用锌粉还原得4-羟基-3-甲氧基-β-氨基乙苯,氢溴酸高温水解得氢溴酸多巴胺,再用盐酸置换得盐酸多巴胺,合成路线如下所示:;上述合成工艺用到的起始原料香兰素为醛类物质,不稳定;硝基甲烷毒性大,为2b类致癌物,易爆炸,为国家管制物质;脱保护用氢溴酸,容易导致成品中含有氢溴酸盐,影响产品质量,且工艺路线长,收率低。
4.路线二(journal of the american chemical society, vol. 133, # 18 p. 6948
ꢀ–ꢀ
6951;indian journal of chemistry, section b: organic chemistry including medicinal chemistry, 1980 , vol. 19, # 9 p. 748
ꢀ–ꢀ
749;journal of the american chemical society, vol. 71, p. 744):以邻苯二甲醚为原料,氯甲基化得
3,4-二甲氧基苄氯,再经氰化、还原得二甲氧基多巴胺,氢溴酸水解得到氢溴酸多巴胺,最后以盐酸置换得盐酸多巴胺合成路线如下所示: ;上述合成工艺同样存在合成路线长、终产品中氢溴酸盐不易除净的缺点,同时还用到剧毒的氰化钾、以及遇氧遇水易爆炸且毒性大的硼烷,不利于工业化生产和劳动保护。
技术实现要素:
5.本发明目的在于克服现有技术缺陷,提供一种无溴盐酸多巴胺合成新方法。该方法采用2-苯甲氧基苯酚为起始原料,经氯甲基化、氰化得4-羟基-3-对苄氧基苯乙腈,再经氢化将氰基还原同时脱去苄基保护并与盐酸成盐得盐酸多巴胺。采用本发明合成方法制备所得盐酸多巴胺具有收率较高(以2-苯甲氧基苯酚计,达50%以上),纯度好(达99.3%以上)等优点。
6.为实现上述目的,本发明采用如下技术方案:一种无溴盐酸多巴胺合成新方法,其以2-苯甲氧基苯酚为起始原料,经卤甲基化、氰化、还原脱保护并与盐酸成盐得盐酸多巴胺,具体包括如下步骤:1)卤甲基化:以2-苯甲氧基苯酚为原料,在卤甲基试剂和溶剂a存在条件下于10~50℃反应4~6小时,获得化合物3;2)氰化:在溶剂b存在条件下,化合物3与氰化试剂反应4-8h,获得化合物4;所述氰化试剂为三甲基氰硅烷、氰化钠或氰化钾;3)还原脱保护并成盐:在乙醇、盐酸和催化剂存在条件下,化合物4与h2在30~70℃、常压~5mpa下反应至不吸氢(即反应压力2小时内不再变化),经后处理即得;合成路线如下所示:
。
7.具体的,步骤1)中,2-苯甲氧基苯酚的卤甲基化产物可以是苄氯或苄溴,相应的,所述卤甲基试剂可以为甲醛溶液加氯化氢、甲醛溶液加溴化氢、或氯甲基烷基醚,优选为氯甲基甲醚。
8.进一步的,步骤1)中,若用甲醛加氯化氢为卤甲基试剂,溶剂a选用盐酸和氯仿;若用甲醛加溴化氢为卤甲基试剂,溶剂a选用氢溴酸和氯仿。若用氯甲基烷基醚为卤甲基试剂,溶剂a选用氯仿,适宜在ph值约为3-4的酸性环境下进行。
9.具体的,步骤1)中,所述卤甲基试剂为甲醛加氯化氢、或甲醛加溴化氢时,2-苯甲氧基苯酚(卤甲基化底物)与甲醛摩尔比为1:1.5~5,优选为1:2.5。所述卤甲基试剂为氯甲基烷基醚时,2-苯甲氧基苯酚与氯甲基烷基醚摩尔比为1:1.05~1.5,优选为1:1.1。
10.进一步的,步骤2)中,氰化反应用的试剂为三甲基氰硅烷、氰化钠、或氰化钾,优选为三甲基氰硅烷。若用三甲基氰硅烷为氰化试剂,溶剂b为二氯甲烷,化合物3与氰化试剂摩尔比为1:1.05~1.5,优选为1:1.125,反应温度为室温。若选用氰化钠或氰化钾为氰化试剂,溶剂b为乙醇,化合物3与氰化试剂摩尔比为1:1.1~1.5,优选为1:1.25,反应温度为乙醇回流温度。
11.进一步的,步骤3)中,氢化反应在乙醇和盐酸的混合溶液中进行,脱去苄基同时将氰基还原为胺基并与盐酸成盐,这将缩短合成路线,降低生产成本,提高反应收率,更加适合工业化。反应中催化剂可选用钯炭催化剂、雷尼镍催化剂、铂催化剂,优选钯炭作为催化剂,反应压力为常压~5mpa,优选为1~2mpa;反应温度为30~70℃,优选为40~50℃。
12.具体的,步骤3)中,催化剂添加量优选为化合物4质量的8-12%。
13.进一步的,步骤3)中,反应至不吸氢后,停止反应,升温到70~80℃,趁热过滤,滤液减压蒸除部分溶剂,降温,析出固体,过滤,得盐酸多巴胺粗品。
14.进一步优选的,盐酸多巴胺粗品用无水乙醇、盐酸回流溶解后,趁热过滤,滤液降温结晶,过滤,减压干燥得盐酸多巴胺成品。
15.和现有方法相比,本发明合成方法具有如下有益效果:1)起始物料用苄醚保护而不用甲醚保护,这样做有两个好处:利用产品合成路线有还原反应特点,将氰基还原和脱保护同时进行,缩短了合成路线;革除了传统工艺脱甲基保护中氢溴酸的使用,产品不存在溴化物的可能性;即便在溴甲基化反应中用到溴化氢,溴
化氢可在后续工艺加碳酸氢钠萃取过程中除去;2)氰化反应传统的氰化试剂氰化钾(钠)有剧毒,本工艺优选采用三甲基氰硅烷作为新型氰化试剂,它具有毒性低、操作安全、适合工业化生产的特点;3)采用本发明合成方法制备所得盐酸多巴胺具有收率较高(以2-苯甲氧基苯酚计,达50%以上),纯度好(达99.3%以上)等优点。
具体实施方式
16.以下结合实施实例对本发明的技术方案作进一步地详细介绍,但本发明的保护范围并不局限于此。
17.实施例中,所用原料均为本领域可直接购买的普通市售产品。室温指代25
±
5℃。
18.卤甲基化:在溶剂a存在条件下,2-苯甲氧基苯酚与卤甲基化试剂反应得到苄氯或苄溴,详见实施例1和2。
19.实施例1
ꢀꢀ
制备4-羟基-3-苄氧基苄氯(化合物3)将35~40%甲醛溶液100g(1.25mol)、37%盐酸200g(2.0mol)、氯仿400g、2-苯甲氧基苯酚(化合物2)100g(0.5mol)投入反应瓶中,连接冷凝管和尾气吸收装置,开启搅拌,缓缓通入氯化氢气体(在反应过程中持续通入,将氯化氢通入反应液面下,能看到冒泡即可,下同),35~45℃保温反应6小时。反应完成后加入水200g,搅拌均匀后,分层,取有机层。水层用氯仿200g萃取一次,合并有机层,饱和碳酸氢钠溶液300ml洗涤、再用水300ml洗涤,减压回收氯仿,得4-羟基-3-苄氧基苄氯100g,收率81%。
20.若要得到4-羟基-3-苄氧基苄溴,将实验中的37%盐酸200g改为68%氢溴酸176g(1.5mol),同时通入氯化氢气体更改为通入溴化氢气体即可。
21.实施例2 制备4-羟基-3-苄氧基苄氯(化合物3)氯仿500g、2-苯甲氧基苯酚100g(0.5mol)投入反应瓶中,连接冷凝管和尾气吸收装置,开启搅拌,通入少量氯化氢气体使反应体系呈酸性(ph值约为3-4),缓缓滴加氯甲基甲醚44.3g(0.55mol),15~25℃保温反应4小时。反应完成后加入水300g,搅拌均匀后,分层,取有机层。水层再用氯仿200g萃取一次,合并有机层,饱和碳酸氢钠溶液300ml洗涤、再用水300ml洗涤,减压回收氯仿,得4-羟基-3-苄氧基苄氯105g,收率85%。
22.实施例3 制备4-羟基-3-苄氧基苯乙腈(化合物4)氮气保护下,三口瓶中加入二氯甲烷500g、4-羟基-3-苄氧基氯苄100g(0.40mol)、三甲基氰硅烷44.7g(0.45mol),开启搅拌,室温反应6小时。反应结束后,缓慢加入硫代硫酸钠溶液300ml(由20g硫代硫酸钠溶于300ml水中制得),搅拌30分钟以破坏多余的氰化物,反应液过滤后分去水层,有机层经水洗、无水硫酸钠干燥后减压蒸去二氯甲烷得粗品,无水乙醇重结晶后,得4-羟基-3-苄氧基苯乙腈80g,收率83%。
23.实施例4:制备4-羟基-3-苄氧基苯乙腈(化合物4)对比氰化反应:氮气保护下,三口瓶中加入无水乙醇500g、4-羟基-3-苄氧基氯苄100g(0.4mol)、开启搅拌,加入氰化钠24.5g(0.5mol),加热回流6小时。反应完成后减压蒸去乙醇,残留物降至室温,加入水300ml和二氯甲烷300ml,搅拌下加入硫代硫酸钠溶液300ml(由20g硫代硫酸钠溶于300ml水中制得),搅拌30分钟以破坏多余的氰化物,用盐酸调ph到4~6,过滤后分去水层,有机层经水300ml洗、无水硫酸钠干燥后减压蒸去二氯甲烷得粗
品,无水乙醇重结晶后,得4-羟基-3-苄氧基苯乙腈74g,收率77%。
24.实施例5:制备盐酸多巴胺(目标化合物1)将乙醇500ml、盐酸40g(含氯化氢约0.4mol)、10%钯炭催化剂(陕西瑞科新材料股份有限公司,型号:d10h5a)6g、4-羟基-3-苄氧基苯乙腈60g(0.25mol)投入耐酸氢化釜中,密闭,氮气置换后于40~50℃、1~2mpa条件下搅拌进行催化氢化并脱苄基反应,反应液不再吸氢时停止反应(即停止通氢后2小时压力不降低,约需20小时)。升温到70~80℃,趁热过滤,滤液减压蒸除部分溶剂,降温,析出固体,过滤,得盐酸多巴胺粗品34g,收率72%。
25.盐酸多巴胺粗品34g加入无水乙醇200g、36~38%浓盐酸18g,回流溶解后趁热过滤,滤液降温到0℃结晶4小时,过滤,70~80℃减压干燥得白色结晶性固体盐酸多巴胺成品,含量99.3%(中国药典2020年二部)。1h nmr (bruker, 500mhz, dmso, δ): 8.85 (2h, d, br), 8.12 (3h, s), 6.69 (1h, d, j=8hz), 6.4 (1h, d, j=2hz), 6.47 (1h, dd, j=8hz, 2hz), 6.69 (1h, d, j=8hz), 2.89 (2h, t, j=7hz), 2.71 (2h, d, j=7hz)。
13
c nmr (bruker, 125mhz, dmso, δ): 145.76,144.51, 128.45, 119.67, 116.54, 116.29, 40.78, 32.84。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。