1.本发明属于中药提取领域,具体涉及一种从光茎短距翠雀中分离的二萜生物碱型化合物及其提取方法和在制备抗炎药物中的应用。
背景技术:
2.光茎短距翠雀(delphinium forrestii var.viride)是一种毛茛科翠雀属植物,分布广泛,主产于西藏、四川、云南等省。1979年版《中国植物志》记载本属植物在民间作用药用,主治跌打损伤、风湿、牙痛、肠炎等症,另有用作农药,杀虱、蚊和蝇的幼虫。近年来,国内外学者陆续报道了毛茛科翠雀属和乌头属等属植物中二萜生物碱的化学成分及其相应的药理作用。目前从翠雀属中分离得到的二萜生物碱类化合物大部分都是由羟基,甲氧基,乙酰基等结构较为简单的基团进行取代。而从光茎短距翠雀分离得到的化合物,部分含有如2
‑
(2
‑
甲基
‑4‑
氧代喹唑啉
‑
3(4h)
‑
基)苯甲酰氧基等结构较为复杂的基团。已知的二萜生物碱可常被认为具有下述作用中的一种或几种,例如抗炎作用(v.n.yu,t.n.povetieva,n.i.suslov,g.n.zyuz'kov,a.v.krapivin,anti
‑
inflammatory activity of diterpene alkaloids from aconitum baikalense,bulletin of experimental biology and medicine 2014,156(5)665
‑
668.)、镇痛作用(wang,d.p.,lou,h.y.,huang,hao,x..j.,liang,g.y.,yang,a novel franchetine type norditerpenoid isolated from the roots of aconitum carmichaeli debx.with potential analgesic activity and less toxicity,bioorganic and medicinal chemistry letters2012,22(13)4444
‑
4446.)、抗癌作用(m.hazawa;k.wada;k.takahashi;t.mori;n.kawahara,i.kashiwakura,suppressive effects of novel derivatives prepared from aconitum alkaloids on tumor growth,investigational new drugs 2009,27(2)111
‑
119.)、抗心律不齐作用(f.a.h.a.ulubelen,h.k.desai,s.w.pelletier,norditerpenoid and diterpenoid alkaloids from turkish consolida orientalis,journal of natural products 2001,64(6)787
‑
789.、抗菌作用(m.ahmad,w.ahmad,m.ahmad,m.zeeshan,obaidullah,f.shaheen,norditerpenoid alkaloids from the roots of aconitum heterophyllum wall with antibacterial activity,j enzyme inhib med chem 2008,23(6)1018
‑
1022.)。目前对于二萜生物碱的抗炎活性研究仅停留在其作用本身,并没有对其抗炎机制进行更进一步的探讨。为了将光茎短距翠雀的药用价值发挥到最大,对光茎短距翠雀的干燥全草进行了系统的成分研究是目前需要进一步探索的。
技术实现要素:
3.本发明的首要目的是提供一种二萜生物碱型化合物。
4.本发明的第二个目的是提供一种二萜生物碱型化合物的提取方法。
5.本发明的第三个目的是提供一种含有二萜生物碱型化合物的药物组合物。
6.本发明的第四个目的是提供上述二萜生物碱型化合物或该化合物的异构体、该化
合物在药学上可接受的盐或包含该化合物的药物组合物在制备抗炎药物中的应用。
7.为实现上述目的,本发明的技术方案概述如下:
8.一种二萜生物碱型化合物,其为如通式(i),(ii)所示的化合物或该化合物的异构体、或该化合物在药学上可接受的盐;
[0009][0010]
其中:r1,r2,r3,r4,r5为氢,羟基,甲氧基,乙酰氧基,邻氨基苯甲酰氧基或2
‑
(2
‑
甲基
‑4‑
氧代喹唑啉
‑
3(4h)
‑
基)苯甲酰氧基。
[0011]
所述化合物为下述结构式所示化合,以及化合物的异构体、该化合物在药学上可接受的盐;
[0012][0013]
所述的在药学上可接受的盐,包括钠盐、钾盐、氨盐、盐酸盐及硫酸盐。
[0014]
所述的异构体,包括:光学异构体、顺反异构体、外消旋体以及它们的混合物。
[0015]
二萜生物碱型化合物的提取方法,以光茎短距翠雀干燥全草为原料经乙醇提取获得所述二萜生物碱型化合物。
[0016]
具体为:
[0017]
(1)以光茎短距翠雀干燥全草为原料,粉碎后加入原料0.1
‑
1质量倍的体积分数为70%
‑
98%的乙醇室温浸泡1
‑
5次,每次5
‑
10天,将提取液减压浓缩后得到浸膏;
[0018]
(2)将总浸膏分散到其2
‑
6质量倍的水中,用盐酸溶液调节混悬液ph至2
‑
3,依次用石油醚、乙酸乙酯分别进行萃取2
‑
5次后,所得萃取液用氨水调节混悬液ph至9
‑
11,混悬液再用二氯甲烷萃取2
‑
5次得到二氯甲烷层浸膏;
[0019]
(3)二氯甲烷层萃取浓缩液经硅胶柱色谱分离,以体积比为100:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,收集体积比为50:1:0.1、15:1:0.1、10:1:0.1的馏分,依次记为d2、d3和d5;
[0020]
(4)馏分d2、d3和d5浓缩后进一步纯化,得到化合物1~6。
[0021]
所述步骤(4)中对馏分d2、d3和d5的具体分离纯化过程为:
[0022]
馏分d2浓缩后经过硅胶柱色谱分离,以体积比为100:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,收集体积比为10:1:0.1,8:1:0.1的馏分,分别记为d26,d27;
[0023]
馏分d26浓缩后经过硅胶柱色谱分离,以体积比为50:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,收集体积比为5:1的馏分,记为d264;
[0024]
馏分d264浓缩后经制备型hplc色谱,以体积比为75:25的甲醇
‑
水为流动相,进行纯化,得到化合物3,4;
[0025]
馏分d27浓缩后经过硅胶柱色谱分离,以体积比为20:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,收集体积比为5:1:0.1的馏分,记为d274;
[0026]
馏分d274浓缩后经制备型hplc色谱,以体积比为75:25的甲醇
‑
水为流动相,进行纯化,得到化合物2;
[0027]
馏分d3浓缩后经过硅胶柱色谱分离,以体积比为100:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,收集体积比为5:1:0.1,3:1:0.1的馏分,分别记为d37,d39;
[0028]
馏分d37浓缩后经过硅胶柱色谱分离,以体积比为20:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,收集体积比为5:1:0.1的馏分,记为d374;
[0029]
馏分d374浓缩后经制备型hplc色谱,以体积比为80:20的甲醇
‑
水为流动相,进行纯化,得到化合物6;
[0030]
馏分d39浓缩后经过硅胶柱色谱分离,以体积比为20:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,收集体积比为5:1:0.1的馏分,记为d394;
[0031]
馏分d394浓缩后经过硅胶柱色谱分离,以体积比为10:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,收集体积比为5:1:0.1的馏分,记为d3944;
[0032]
馏分d3944浓缩后经制备型hplc色谱,以体积比为75:25的甲醇
‑
水为流动相,进行纯化,得到化合物1;
[0033]
馏分d5浓缩后经反相ods柱色谱分离以体积分数为40%~90%的甲醇
‑
水为洗脱剂进行洗脱,收集体积分数为60%的馏分,记为d52;
[0034]
馏分d52浓缩后经制备型hplc色谱以体积比为75:25的甲醇
‑
水为流动相,进行纯化,得到化合物5。
[0035]
一种药物组合物,组合物含所述二萜生物碱型化合物、该化合物的异构体、该化合物在药学上可接受的盐中的一种或多种。
[0036]
一种药物制剂,制剂为活性成分和药学上可接受的载体、赋形剂、稀释剂中的一种或它们的组合;其中,活性成分为所述化合物或所述组合物。
[0037]
所述制剂的给药途径为口服或注射给药,剂型为:片剂、胶囊剂、粉剂、糖浆剂或针剂。
[0038]
一种二萜生物碱型化合物或药物组合物或药物制剂的应用,所述二萜生物碱型化合物、所述组合物、所述的药物制剂在制备抗炎药物中的应用。
[0039]
所述的二萜生物碱型化合物、该化合物的异构体、该化合物在药学上可接受的盐或所述的药物组合物对lps诱导的raw264.7细胞中no产生的抑制作用,应用于制备抗炎药物。
[0040]
所述二萜生物碱型化合物、所述组合物、所述的药物制剂在作为预防及治疗类风湿性关节炎,炎症性肠病或动脉粥样硬化的药物中的应用。
[0041]
本发明所具有的优点:
[0042]
本发明所得二萜生物碱型化合物由光茎短距翠雀提取获得,利用核磁、质谱等手段对化合物的结构确证可见从光茎短距翠雀中分离得到的部分二萜生物碱含有如2
‑
(2
‑
甲基
‑4‑
氧代喹唑啉
‑
3(4h)
‑
基)苯甲酰氧基等结构较为复杂的基团;同时所得二萜生物碱型化合物或其异构体或其在药学上可接受的盐或其药物组合物对lps诱导的raw264.7细胞中no产生的抑制作用,并且通过抑制ros的产生并调节nf
‑
κb、mapk和nrf2信号通路起到抗炎作用,应用于制备治疗炎症的药物。本发明进一步丰富光茎短距翠雀活性物质的结构多样性,在此基础上,为后续得到的单体化合物进行相关生物活性测试奠定基础,为新药开发提供活性先导化合物,同时也为光茎短距翠雀药材的深层次研究与开发提供理论依据。
附图说明
[0043]
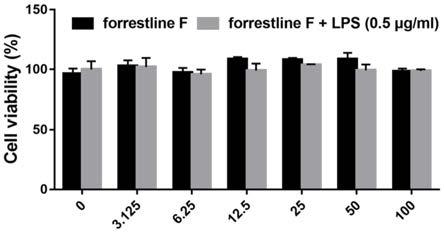
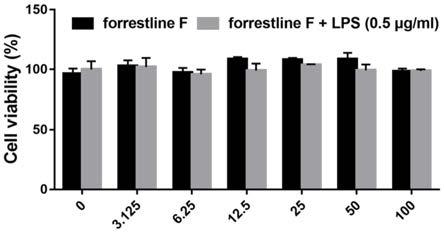
图1不同浓度的化合物6在lps存在或不存在的情况下对raw264.7细胞的存活率效果图。
[0044]
图2化合物6对lps诱导的巨噬细胞raw264.7分泌的炎症细胞因子的表达效果图。(a)(b)(c)图示elisa试剂盒检测细胞上清中炎症因子il
‑
1β、tnf
‑
α和il
‑
6的表达。
###
p<0.001vs对照组,***p<0.001vs lps组。
[0045]
图3化合物6对inos、cox
‑
2以及nf
‑
κb p65、mapks、nrf2信号通路相关蛋白的表达情况效果图。(a)(b)(c)图示western
‑
blot检测炎症因子inos、cox
‑
2蛋白水平的表达及对应的灰度统计图。(d)图示western
‑
blot检测p
‑
p65、p65、p
‑
iκbα、iκbα的表达,(e)图为(d)图的灰度分析情况,显示p
‑
p65、p
‑
iκbα蛋白的相对表达量。(g)图示western
‑
blot检测p
‑
p38、p38、p
‑
erk、erk、p
‑
jnk、jnk的表达,(f)图为(g)图的灰度分析情况,显示p
‑
p38、p
‑
erk和p
‑
jnk蛋白的相对表达量。(h)(i)图示western
‑
blot检测keap
‑
1、nrf2、ho
‑
1的蛋白表达以及对应的灰度统计图。
###
p<0.001vs对照组,**p<0.01vs lps组,***p<0.001vs lps组。
[0046]
图4化合物6对nf
‑
κb p65、nrf2核转位的影响效果图。(a)图示化合物6对lps诱导的nf
‑
κb p65核转位的抑制效应,蓝色荧光为dapi标记的核,红色荧光为nf
‑
κb p65。(b)图示化合物6对lps诱导的nfr2核转位的促进效应,蓝色荧光为dapi标记的核,红色荧光为nrf2。
[0047]
图5化合物6对活性氧以及线粒体膜电位水平的影响效果图。(a)图示除对照组和lps组外,细胞用化合物6不同浓度处理3小时后再加入lps,作用12h。收集细胞,经dcfh
‑
da染色后通过荧光显微镜获得图像。(b)图示细胞按上述方法处理,用流式细胞术检测dcfh
‑
da的荧光强度。(c)通过flow jo软件计算的dcfh
‑
da荧光强度。(d)(e)图示通过流式细胞术检测线粒体膜电位水平,并计算jc
‑
1两种状态下的细胞比例。
###
p<0.001vs对照组,**p<
0.01vs lps组,***p<0.001vs lps组。
具体实施方式
[0048]
下面将结合具体实施例对本发明的技术方案进行进一步描述。
[0049]
实施例1
[0050]
光茎短距翠雀中二萜生物碱型化合物1~6的提取方法:
[0051]
(1)以总干重为10.0kg的光茎短距翠雀为原料,加入原料0.4质量倍的95%乙醇试剂(4l),室温浸泡3次,每次7天。将提取液减压浓缩后得到浸膏(360g)。
[0052]
(2)将总浸膏分散到其2质量倍的水(720ml)中,用盐酸溶液调节混悬液ph至2,依次分别用石油醚、乙酸乙酯进行萃取3次后,用氨水调节混悬液ph至10,并用二氯甲烷萃取3次得到二氯甲烷层浸膏(75g)。
[0053]
(3)二氯甲烷层萃取浓缩液干法拌样后经200
‑
300目硅胶柱色谱分离,以体积比为100:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,洗脱速度为1.2ml/min,收集体积比为50:1:0.1、15:1:0.1、10:1:0.1的馏分,依次记为d2、d3和d5;
[0054]
(4)馏分d2、d3和d5减压浓缩后进一步纯化,得到27.2mg化合物1、55mg化合物2、235mg化合物3、53mg化合物4、32.9mg化合物5和10.7mg化合物6。具体分离纯化过程为:
[0055]
馏分d2经减压浓缩后得到22.6g浓缩液,干法拌样后经200
‑
300目硅胶柱色谱分离,以体积比为50:1:0.1、15:1:0.1、10:1:0.1、8:1:0.1、5:1:0.1、3:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,洗脱流速为2.5ml/min,收集体积比为10:1:0.1,8:1:0.1的馏分,分别记为d26,d27;
[0056]
馏分d26经减压浓缩后得到2.6g浓缩液,干法拌样后经200
‑
300目硅胶柱色谱分离,以体积比为50:1:0.1、40:1:0.1、30:1:0.1、20:1:0.1、15:1:0.1、10:1:0.1、5:1:0.1、2:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,洗脱流速为2.5ml/min,收集体积比为5:1:0.1的馏分,记为d264;
[0057]
馏分d264浓缩后得到1.7g,经制备型hplc色谱,色谱柱条件为shimadzu 5μm c18 20
×
250mm,以体积比为75:25的甲醇
‑
水为流动相,流速为8ml/min,进行纯化,得到235mg化合物3,53mg化合物4;
[0058]
馏分d27浓缩后得到1.9g浓缩液,干法拌样后经200
‑
300目硅胶柱色谱分离,以体积比为50:1:0.1、40:1:0.1、30:1:0.1、20:1:0.1、15:1:0.1、10:1:0.1、5:1:0.1、2:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,洗脱流速为2.5ml/min,收集体积比为5:1:0.1的馏分,记为d274;
[0059]
馏分d274浓缩后得到335mg,经制备型hplc色谱,色谱柱条件为shimadzu 5μm c18 20
×
250mm,以体积比为75:25的甲醇
‑
水为流动相,流速为8ml/min,进行纯化,得到55mg化合物2;
[0060]
馏分d3浓缩后得到3g浓缩液,干法拌样后经200
‑
300目过硅胶柱色谱分离,以体积比为50:1:0.1、30:1:0.1、15:1:0.1、10:1:0.1、5:1:0.1、3:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,洗脱流速为2.5ml/min,分别收集体积比为5:1:0.1,3:1:0.1的馏分,记为d37,d39;
[0061]
馏分d37浓缩后得到2.1g浓缩液,干法拌样后经200
‑
300目硅胶柱色谱分离,以体
积比为50:1:0.1、40:1:0.1、30:1:0.1、20:1:0.1、15:1:0.1、10:1:0.1、5:1:0.1、2:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,洗脱流速为2.5ml/min,收集体积比为5:1:0.1的馏分,记为d374;
[0062]
馏分d374浓缩后得到40mg,经制备型hplc色谱,色谱柱条件为shimadzu 5μm c18 20
×
250mm,以体积比为75:25的甲醇
‑
水为流动相,流速为8ml/min,进行纯化,得到10.7mg化合物6;
[0063]
馏分d39浓缩后得到1.1g浓缩液,干法拌样后经200
‑
300目硅胶柱色谱分离,以体积比为20:1:0.1、15:1:0.1、10:1:0.11、8:1:0.1、5:1:0.1、2:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,洗脱流速为2ml/min,收集体积比为5:1:0.1的馏分,记为d394;
[0064]
馏分d394浓缩后得到1.1g浓缩液,干法拌样后经200
‑
300目硅胶柱色谱分离,以体积比为15:1:0.1、10:1:0.11、8:1:0.1、5:1:0.1、2:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,洗脱流速为2ml/min,收集体积比为5:1:0.1的馏分,记为d3944;
[0065]
馏分d3944浓缩后得到320mg,经制备型hplc色谱,色谱柱条件为shimadzu 5μm c18 20
×
250mm,以体积比为75:25的甲醇
‑
水为流动相,进行纯化,流速为8ml/min,得到27.2mg化合物1;
[0066]
馏分d5浓缩后得到10.5g经反相ods柱色谱分离以体积分数为40%、60%、70%、90%的甲醇
‑
水为洗脱剂进行洗脱,收集体积分数为60%的馏分,记为d52。
[0067]
馏分d52浓缩后得到经制备型hplc色谱,色谱柱条件为shimadzu5μm c18 20
×
250mm,以体积比为75:25的甲醇
‑
水为流动相,进行纯化,得到32.9mg化合物5。
[0068]
化合物的物理化学和常数如下:
[0069]
化合物1:白色无定型粉末;hresims m/z:670.3463[m h]
(calcd for c
39
h
48
n3o7,670.3487),确定化合物1的分子式为c
39
h
47
n3o7;
‑
202.0(c 0.3,meoh);1h
‑
nmr(600mhz,cdcl3)和
13
c
‑
nmr(150mhz,cdcl3),数据见表1。
[0070]
化合物2:白色无定型粉末;hresims m/z:684.3623[m h]
(calcd for c
40
h
50
n3o7,684.3643),确定化合物2的分子式为c
40
h
49
n3o7;
‑
143.0(c 0.3,meoh);1h
‑
nmr(600mhz,cdcl3)和
13
c
‑
nmr(150mhz,cdcl3),数据见表2。
[0071]
化合物3:白色无定型粉末;hresims m/z:698.3767[m h]
(calcd for c
41
h
51
n3o7,698.3799),确定化合物3的分子式为c
41
h
50
n3o7;
‑
331.5(c 0.4,meoh);1h
‑
nmr(600mhz,cdcl3)和
13
c
‑
nmr(150mhz,cdcl3),数据见表3。
[0072]
化合物4:白色无定型粉末;hresims m/z:569.3210[m h]
(calcd for c
32
h
45
n2o7,569.3221),确定化合物4的分子式为c
32
h
44
n3o7;
‑
332.0(c 0.2,meoh);1h
‑
nmr(600mhz,cdcl3)和
13
c
‑
nmr(150mhz,cdcl3),数据见表4。
[0073]
化合物5:白色无定型粉末;hresims m/z:436.3045[m h]
(calcd for c
25
h
42
no5,436.3058),确定化合物5的分子式为c
25
h
41
no5;
‑
141.0(c 0.5,meoh);1h
‑
nmr(600mhz,cdcl3)和
13
c
‑
nmr(150mhz,cdcl3),数据见表5。
[0074]
化合物6:白色无定型粉末;hresims m/z:316.2261[m h]
(calcd for c
20
h
30
no2,316.2271),确定化合物6的分子式为c
20
h
29
no2;
‑
468.5(c 0.1,meoh);1h
‑
nmr(600mhz,
cdcl3)和
13
c
‑
nmr(150mhz,cdcl3),数据见表6。
[0075]
表1化合物1的碳谱和氢谱数据
[0076][0077]
注:1h
‑
nmr,600mhz,cdcl3;
13
c
‑
nmr,150mhz,cdcl3。
[0078]
表2化合物2的碳谱和氢谱数据
[0079]
[0080]
注:1h
‑
nmr,600mhz,cdcl3;
13
c
‑
nmr,150mhz,cdcl3。
[0081]
表3化合物3的碳谱和氢谱数据
[0082][0083]
注:1h
‑
nmr,600mhz,cdcl3;
13
c
‑
nmr,150mhz,cdcl3。
[0084]
表4化合物4的碳谱和氢谱数据
[0085][0086]
注:1h
‑
nmr,600mhz,cdcl3;
13
c
‑
nmr,150mhz,cdcl3。
[0087]
表5化合物5的碳谱和氢谱数据
[0088][0089]
注:1h
‑
nmr,600 mhz,cdcl3;
13
c
‑
nmr,150 mhz,cdcl3。
[0090]
表6化合物6的碳谱和氢谱数据
[0091][0092]
注:1h
‑
nmr,600 mhz,cdcl3;
13
c
‑
nmr,150 mhz,cdcl3。
[0093]
通过理化常数和现代波谱学手段(hresims和nmr),结合文献相关数据,鉴定它的结构,化合物1、2、3、4、5和6结构,如下所示:
[0094][0095]
实施例2
[0096]
(1)以总干重为15.0kg的光茎短距翠雀为原料,加入原料0.3质量倍的95%乙醇试剂(4.5l),室温浸泡3次,每次6天。将提取液减压浓缩后得到浸膏(400g),
[0097]
(2)将总浸膏分散到2质量倍的水(800ml)中,用盐酸溶液调节混悬液ph至2.5,依次用石油醚乙酸乙酯进行萃取3次后,用氨水调节混悬液ph至10.5,并用二氯甲烷萃取得到二氯甲烷层浸膏(81g)。
[0098]
(3)二氯甲烷层萃取浓缩液经硅胶柱色谱分离,以体积比为100:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,收集体积比为50:1:0.1、15:1:0.1、10:1:0.1的馏分,依次记为d2、d3和d5;
[0099]
(4)馏分d2、d3和d5浓缩后进一步纯化,得到29.2mg化合物1、61mg化合物2、272mg化合物3、61mg化合物4、34.9mg化合物5和10.8mg化合物6。
[0100]
实施例3
[0101]
以总干重为12.0kg的光茎短距翠雀为原料,加入原料0.2质量倍的95%乙醇试剂(2.4l),室温浸泡3次,每次7天。将提取液减压浓缩后得到浸膏(350g)。
[0102]
(2)将总浸膏分散到2质量倍的水(700ml)中,用盐酸溶液调节混悬液ph至3,依次用石油醚乙酸乙酯进行萃取3次后,用氨水调节混悬液ph至11,并用二氯甲烷萃取得到二氯甲烷层浸膏(77g)。
[0103]
(3)二氯甲烷层萃取浓缩液经硅胶柱色谱分离,以体积比为100:1:0.1~0:1:0.1的石油醚
‑
丙酮
‑
二乙胺为洗脱剂梯度洗脱,收集体积比为50:1:0.1、15:1:0.1、10:1:0.1的馏分,依次记为d2、d3和d5;
[0104]
(4)馏分d2、d3和d5浓缩后进一步纯化,得到28.2mg化合物1、60mg化合物2、265mg化合物3、60mg化合物4、34.1mg化合物5和10.2mg化合物6。
[0105]
实施例4
[0106]
本发明产物对raw264.7细胞生成no的影响研究
[0107]
将raw264.7细胞接种在96孔板中,用20μm的上述实施例1制备获得化合物处理3小时,然后与lps(0.5μg/ml)一起孵育24小时。将具有或不具有lps的dmso作为媒介物对照或
模型处理。使用griess试剂在540nm下用酶标仪测量培养基中的亚硝酸盐积累。计算化合物处理组的抑制率(%)以评价no抑制活性。
[0108]
表1.化合物抑制raw264.7细胞no生成抑制率(%)值表
[0109]
化合物抑制率(%)化合物112.44化合物237.20化合物310.30化合物699.14
[0110]
从上述表中可以发现,化合物6有较强的抑制活性。
[0111]
进一步的对上述化合物6在raw264.7细胞中抗炎作用与机制研究
[0112]
(1)cck8法检测化合物6对细胞存活率的影响
[0113]
对数生长期的raw264.7细胞以25000个/孔接种于96孔板中培养12h。换不同浓度的化合物6(100、50、25、12.5、6.25和3.125μmol/l)处理细胞。以加入相应体积的dmso的细胞孔作为空白对照。24h后弃掉培养基,每孔均加入100μl的含10%cck8的培养基,培养40min后,酶标仪检测450nm处各孔的od值计算实验组细胞存活率(以空白对照组细胞存活率为100%)。结果图1所示:
[0114]
由图1可知,化合物6对raw264.7细胞的存活率没有显著影响。
[0115]
(2)elisa法检测化合物6对白介素1β(il
‑
1β)、白介素6(il
‑
6)和tnf
‑
α的生成的抑制作用
[0116]
对数生长期的raw264.7细胞接种于96孔板培养12h。实验组用不同浓度化合物6(5、10、20和40μmol/l)处理。3h后,模型组和实验组加入lps,使其终浓度为0.5μg/ml,而空白对照组加入等体积的dmem培养基,继续培养24h。每孔均取细胞上清,按照elisa试剂盒操作手册,于酶标仪检测450nm处各孔的od值,计算每组的细胞因子含量。结果如图2所示:
[0117]
由图2可知,化合物6可以抑制lps引起的巨噬细胞raw264.7分泌的细胞因子含量,并且,其抑制作用呈现剂量依赖性。
[0118]
(3)western blot检测化合物6对炎症相关蛋白以及nf
‑
κb、mapk、nrf2/ho
‑
1通路表达的抑制作用
[0119]
raw264.7细胞接种于96孔板培养12h。实验组用不同浓度化合物6(5、10和20μmol
·
l
‑1)处理。3h后,模型组和实验组加入lps,使其终浓度为0.5μg/ml,而空白对照组加入等体积的dmem培养基,继续培养24h。弃掉培养基,收集细胞,用ripa裂解,bca定量进行western blot实验,检测不同浓度化合物6处理后inos、cox
‑
2以及nf
‑
κb、mapks、nrf2信号通路相关蛋白的表达情况,曝光条带用gel
‑
pro analyzer进行灰度分析。结果如图3所示。
[0120]
由图3可知,化合物6可以通过降低inos、cox
‑
2、nf
‑
κb及mapks通路和上调nrf2信号通路及下游相关蛋白的表达而发挥抗炎作用。
[0121]
(4)免疫荧光法检测化合物6对nf
‑
kb p65、nrf2核转位的影响
[0122]
将raw264.7细胞接种到每孔8
×
104细胞的24孔板中,培养12h,然后用dmso或6(20μm)预处理2h,并用0.5μg/ml lps刺激12h。用新鲜制备的4%多聚甲醛固定细胞10min,用pbs洗涤3次,接着用0.2%triton x
‑
100通透10分钟。在室温下用5%牛血清白蛋白(bsa)封闭1h后,加入1:400稀释的nf
‑
κb p65(proteintech,cat#10745
‑1‑
ap)抗体或nrf2
(proteintech,cat#16396
‑1‑
ap),并在4℃下孵育过夜。通过pbs洗涤后,在室温下和暗处以1:400的稀释度添加二抗孵育1h。最后,在室温和暗处下用dapi染色5min。然后pbs清洗并加入抗荧光淬灭封片液,在免疫荧光显微镜下观察并拍照,并获取图像。结果如图4所示。
[0123]
由图4可知,化合物6(20μm)可以显著抑制lps
‑
激活的raw264.7细胞中nf
‑
κb的p65亚基从胞浆转移到细胞核,同时,化合物6(20μm)可以促进lps诱导的nrf2核易位。
[0124]
(5)化合物6对活性氧水平以及线粒体膜电位水平的影响
[0125]
收集并离心lps诱导的raw264.7细胞,离心后,用pbs清洗细胞三次。然后,按照试剂盒说明书的操作步骤,加入dcfh
‑
da或jc
‑
1荧光染料,在37℃下染色30min,收集染色细胞并使用流式细胞仪或荧光显微镜进行检测分析,结果如图5所示。
[0126]
由图5可知,化合物6可剂量依赖性的抑制活性氧及线粒体膜电位水平。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。