far1基因的新用途
技术领域
1.本发明属于医药技术领域,具体涉及far1基因的新用途。
背景技术:
2.非小细胞肺癌(nsclc)占肺癌总数超过 80%,与吸烟习惯、环境污染密切相关。肺癌是癌症病症头号杀手,每年全球都有超过一百万的病人因此丧生。肺癌发生率和死亡率互相紧密联系,尽管诊断技术和治疗方法在不断改进创新,但肺癌的总体 5年生存率依旧很低,全球形势不容乐观。肿瘤发病机制复杂多变,肿瘤的靶向治疗在诊断及治疗 nsclc中始终占据关键地位。因此,深入探究肺癌细胞信号传导网络,研究可靠的治疗靶点是治疗nsclc的一个重要途径。
3.mapk信号通路的异常表达和过度激活是导致癌症发生发展的重要通路,已经有许多靶向mapk信号通路的药物进行临床试验,并且有部分药物,如靶向b-raf的抑制剂dabrafenib以及靶向mek的抑制剂trametinib已经被fda批准在临床联合使用治疗nsclc,不同于该通路上的其他“明星”蛋白,a-raf蛋白由于激酶活性较弱等原因,对其在抗肿瘤的研究较少,但是在肺腺癌患者中也发现了a-rafs241c突变,因此通过研究a-raf结合蛋白,阐明他们之间的调控机制,可以为nsclc的治疗提供新的靶点。raf是细胞内信号通路mapk上的枢纽蛋白,包括调控细胞的增殖、分化、迁移和凋亡等很多生物学过程,这条信号通路的异常激活,造成细胞内生物学功能的紊乱,最终导致肿瘤的发生。
4.far1是一种过氧化物酶体膜蛋白,参与醚脂和甘油磷脂等脂质的合成,far1优先将c16和c18的磷脂酰辅酶a还原为脂肪醇,该步骤是醚脂合成的限速步骤,为有机体合成醚脂/缩醛磷脂、蜡单脂提供底物。醚脂和甘油磷脂等脂质参与调控细胞生物学功能及一系列信号转导,当细胞内的脂质稳态发生变化时会导致细胞癌变。已有研究指出,当小鼠暴露于pm
2.5
的环境中时,体内甘油三酯、游离脂肪酸水平增加,并伴随缩醛磷脂紊乱,使肺部更容易引发肺炎,进一步促进肺癌的发生。但是对在癌症发展过程中far1蛋白在其中发挥的功能的相关研究却很少,其涉及的分子机制尚不明确,far1作为a-raf激酶的结合蛋白之一,可以成为far1蛋白功能研究的新方向。
5.靶向治疗是目前临床对nsclc患者比较有效的治疗手段之一,通过靶向目的基因/蛋白,改变癌细胞内信号通路,阻止癌变的发展从而达到治疗效果,因此探索nsclc的治疗靶点具有重要意义。
技术实现要素:
6.本发明提供了far1基因的新用途,即以抑制far1基因表达为目的的筛选用于治疗非小细胞肺癌药物的应用,所述far1基因的核苷酸序列见genebank中基因登录号为id:84188。
7.异常的脂质代谢是癌细胞的特征之一,特别是醚脂和磷脂在肿瘤中的变化,但是他们在癌症中的具体功能扔不清楚。far1蛋白作为缩醛磷脂合成途径的限速酶,为了深入
了解far1蛋白的功能和基质,首先要了解far1蛋白参与调控的脂质,因此利用crispr/cas9技术敲除a549细胞中的far1基因通过代谢组学比较far1基因敲除的a549细胞与野生型a549细胞之间细胞内脂质含量的差异。通过比较差异脂质的归类分析,了解far1所参与的脂质代谢途径,有利于更加全面得了解far1蛋白的生物学功能。
8.本实验成功构建敲除far1基因的a549细胞株a549-ko-far1(akf),通过非靶向脂质代谢组学分析,在正常肺泡上皮细胞(hpaepic)和肺腺癌细胞(a549)之间共有97个差异脂质,其中c16和c18的脂质有47个,差异脂质主要类别为甘油三酯(tg)、磷脂酰胆碱(pc)、磷脂酰乙醇胺(pe)。通过非靶向脂质代谢组学分析发现,在a549-ko-far1(akf)和野生型a549之间共有102个差异脂质,其中c16和c18的脂质有51个,差异脂质主要类别为甘油三酯(tg)、磷脂酰胆碱(pc)、磷脂酰乙醇胺(pe)。通过两两比较,发现敲除far1基因后,a549中部分tg和pc脂质水平得到恢复,并且qpcr的结果也证明了这一点,表明far1调控a549细胞中的脂质代谢。通过敲除a549细胞中的far1基因开展功能试验发现far1蛋白能够促进肺腺癌细胞增殖和迁移的能力,进而说明可以通过抑制far1蛋白表达来达到治疗非小细胞肺癌的目的,本发明为将来基于far1蛋白的非小细胞肺癌治疗药物的开发提供了可能,本发明具有较大的应用价值和前景。
附图说明
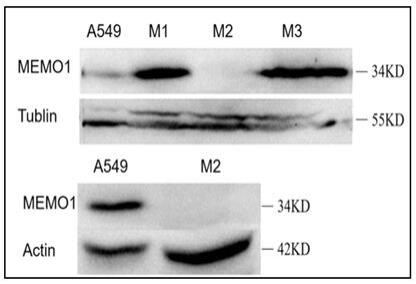
9.图1为western blotting验证a549细胞稳定敲除far1单克隆细胞株结果示意图;图2为px459-far1基因敲除细胞系pcr扩增片段序列比对结果示意图;图3为正常肺泡上皮细胞、肺腺癌细胞a549和akf 细胞的c16和c18差异脂质统计分析热图;图4为三种细胞c16和c18差异脂质分析柱状图;图5 为三种细胞c16和c18差异脂质分析柱状图;图6 为两组样品c16和c18差异脂质表达结果韦恩示意图;图7 为参与pc合成与代谢的蛋白的qpcr分析;图中*p《0.05,** p《0.01,*** p《0.001,**** p《0.0001,t-test;图8 为参与tg合成与代谢的蛋白的qpcr分析;图中*p《0.05,** p《0.01,*** p《0.001,**** p《0.0001,t-test;图9 为细胞增殖实验结果示意图,图中 *** p《0.001,t-test;图10 为细胞克隆形成实验结果,图中*p《0.05, t-test,其中上图为克隆培养结果,下图为统计数据;图11 为细胞划痕实验结果;图中*** p《0.001,t-test,其中上图为细胞划痕实验结果,下图为统计数据;图12 为细胞transwell迁移实验结果; *** p《0.001,**** p《0.0001,t-test,其中上图为迁移实验结果,下图为统计数据;上述图例中akf即为a549-far1-ko稳转细胞株,即稳定敲除far1的a549细胞株;rescue为在敲除far1的细胞中转入far1高表达质粒,属于恢复实验,作为对照组。
具体实施方式
10.下面通过实施例来进一步说明本发明的实质性内容,但本发明的内容并不局限于此,本实施例中方法如无特殊说明均为常规方法,所用试剂如无特殊说明,均为常规时售试剂或按常规方法配制的试剂。
11.实施例1:构建far敲除质粒 crispr/cas9是细菌和古细菌在长期的演化过程中形成的一种适应性免疫防御,用来抵御入侵病毒和外源dna。微生物将入侵的dna序列整合到自身的基因组中,当再次收到入侵时,通过切割病毒等的dna或rna进行切割,从而抵御病毒的侵害。近几年crispr/cas9被人源化用于基因编辑,通过对靶向基因进行特定的dna修饰,对疾病的治疗提供新的方案。crispr (clusteredregularly interspaced short palindromic repeat)在编辑基因的时候需要两个部分:cas9核酸酶和grna,其中cas9核酸酶已经被整合到商业化的载体中,而grna则是crispr敲除系统中重要的组成部分。crispr序列中有靶向定位于目的基因的20个nt左右的 rna序列,cas9和grna 结合形成cas9核糖核蛋白(ribonucleoprotein,rnp),rnp定位到目的基因的靶序列上,对目的基因进行切割,使dna链断裂受到损伤,利用细胞倾向于非同源重组的修复造成移码突变,使目的基因发生突变,提前终止或者丧失原有功能。
12.1、设计靶向far1基因的sgrna以及验证引物利用在线设计平台http://chopchop.cbu.uib.no/设计靶向far1基因的sgrna,选择h. sapiens和crispr/cas9后点击find target sites。根据排序选择靠目标位置位于上游的外显子的sgrna序列,本实验选择了两组sgrna片段,见下表:表1:靶向far1基因的sgrna片段序列注:小写序列为sgrna与pspcas9(bb)-2a-puro(px459)质粒连接的粘性末端;将每条sgrna溶解至终浓度为100μm,按照表2的退火反应体系对sgrna进行磷酸化和退火;表2:磷酸化和退火体系;将上述混合物放置pcr仪中,设置程序:37℃,30min;95℃,5min,然后以5℃/min降
至25℃,对sgrna进行磷酸化和退火形成双链;将上述已经磷酸化和退火的产物按1:200的比例用无rna酶水进行稀释,按照以下配方将上述稀释的产物与px459质粒进行连接;表3:连接体系将配置好上述混合物置于pcr仪中,按37℃,5min;21℃,5min的程序进行6个循环反应;然后水解未连接成功的线性dna,水解体系如下:表4:水解线性dna体系;将配制好的反应体系置于pcr仪,水解条件为:37℃,30 min;70℃,30min,然后取7.5μl水解产物进行大肠杆菌dh5α转化实验,在含有氨苄霉素(ampicillin,amp)的固体lb培养基上挑取单克隆,摇菌后送去测序(测序引物为u6启动子通用引物),保留测序结果正确的质粒进行后续实验。
13.实施例2:构建far1基因敲除稳定细胞株1、选取代数靠前,并且状态良好的a549细胞接种于6孔板中,保证24h后细胞密度为75%左右,使用只含有10%胎牛血清的rpmi-1640培养基在37℃、5%的co2细胞培养箱中培养;2、转染质粒前,先用无菌1
×
pbs清洗一遍细胞,加入optim培养基,每孔转染质粒1.2μg,另外转染未链接sgrna的px459质粒作为阴性对照(在保持转染质粒总质量为1.2μg的前提下,将两组sgrna混合在一起转染到a549细胞中),转染6h后,将培养基换为含有血清但是不含抗生素的完全培养基继续培养;3、转染24h后,将细胞传入100mm的细胞皿中继续培养,待第二天加入0.8μg /ml的嘌呤霉素继续培养,筛选阳性单克隆,筛选期间,每隔4天换液一次,直至有阳性克隆出现;4、15天后,配合使用克隆杯将阳性克隆转移到12孔板中继续培养;
5、待细胞长满后传代至6孔板中,待6孔板中的细胞每孔覆盖率达80%后即可用与western blot和pcr测序检测所选细胞克隆的基因敲除情况;将western blotting和测序结果都为阳性的细胞克隆扩大培养并保存;a、细胞总蛋白提取细胞收样:6孔板中的单克隆细胞弃培养基,用4℃预冷的1
×
pbs清洗一次,放在冰上;细胞裂解:向六孔板内每孔加入100μl的1
×
sds-page loading buffer(加入β巯基乙醇),在涡旋震荡仪上震荡15s,促进细胞总蛋白的提取,冰上裂解30 min;蛋白线性化:98℃金属浴5 min进行蛋白变性,待样品冷却后-20℃保存。
14.b、用western bolt法检测单克隆细胞株的基因敲除情况将玻璃板清洗干净后再用去离子水冲洗一遍,晾干待用;配胶:配制10%的分离胶(下层胶)和5%的浓缩胶(上层胶);上样:将凝胶板放入电泳槽后,倒入适量的l
×
电泳缓冲液,将制胶梳小心拔去,将蛋白mark以及置备好的蛋白样品依次加入sds-page凝胶的各泳道中;电泳:将电泳条件设置为80v恒压,时间为30min左右,样品到达下层胶后,设置电泳条件为120v,1h10min,当溴酚蓝跑到凝胶底部时,可终止工作。
15.转膜:将预先配置好的转膜液放入4℃预冷,转膜采用三明治-湿转方法;转膜放置顺序依次为棉垫-三张滤纸(6
×
8cm)-凝胶(分离胶)-pvdf膜(已用无水甲醇激活)-三张滤纸-棉垫,全程要避免气泡,将夹子扣紧后放入转膜槽中,加入适量转膜缓冲液;转膜条件为:110v,1h 35min,在转膜期间将转膜槽置于冰水混合液中,用以中和转膜时产生的热量;封闭:转膜结束后,将pvdf膜浸泡在5%的脱脂牛奶封闭溶液中,室温,摇床封闭0.5-1h;孵育一抗:用2%的脱脂牛奶( l
×
tbst配制)按比例稀释抗体,将pvdf膜放入抗体盒中,放入配置好的一抗,保证抗体可以完全覆盖pvdf膜,在4℃冷库,摇床孵育过夜;孵育二抗:将pvdf膜用1
×
tbst清洗三次,每次5min,将清洗好的膜放入抗体盒后加入对应的二抗,保证抗体可以完全覆盖pvdf膜,室温,摇床孵育50min;化学发光仪显影:将pvdf膜用1
×
tbst清洗三次,每次5min,将配置好的显影液覆盖在pvdf膜上,放入化学发光仪中显影;western blotting验证结果见图1,结果显示将构建好的两个质粒混合进行稳转,用嘌呤霉素筛选单克隆株,获得构建成功的a549-ko-far1细胞株(akf)。
16.c、将western blot验证成功的细胞株进行基因测序,在基因水平验证far1基因的敲除情况待测细胞全基因组的提取操作按照常规市售动物基因组dna提取试剂盒的说明书进行;设计验证引物:上游引物-f:atcaaaatggtttcaatcccag,下游引物-r:
gacttcttccactcgctcttgt,所扩增的片段涵盖了sgrna打靶的位点;按照表5制备pcr反应体系表5:pcr反应混合体系
④
pcr扩增反应条件如下:将获得的产物送测序并将敲除组的序列与野生型a549细胞序列比对,将成功基因敲除的细胞记为akf;pcr扩增片段序列比对结果见图2,表示已经成功获得稳定敲除far1基因的a549细胞系。
17.实施例3:非靶向脂质代谢组学检测实验1)准备细胞:将状态良好,细胞密度为95%左右的肺腺癌细胞a549、akf细胞、正常肺泡上皮细胞hpaepic(保证每皿的细胞量≥107个细胞)的培养基去除,用1
×
pbs清洗两次后,用细胞刮将细胞收集到2ml的离心管中,离心去上清,将细胞沉淀保存在液氮中;2)取100mg样本置于5ml离心管中,加入1.5ml氯仿-甲醇-水混合液(2:2:1,-20℃),加入5颗钢珠;3)将样品放入高通量组织研磨仪中,60hz处理1.5min;4)将样品至于冰上静置30min,加入0.38ml纯水,涡旋震荡30s,置于冰上静置10min;5)室温下,12000r离心5min,取600μl下层液于新的2ml离心管中;
6)加入1ml氯仿-甲醇-水的混合溶液(2:2:1,-20℃),涡旋震荡30s;7)室温下,12 000r离心5min,取800μl下层液于新的2ml离心管中,用真空浓缩仪浓缩样品;8)用200μl异丙醇将样品溶解后,0.22μm膜过滤样品后,每个待测样本取20μl混合成qc样本(用于矫正混合样品分析结果的偏差以及由于分析仪器自身原因造成的失误),剩余样品进行lc-ms检测;色谱条件:仪器采用thermo ultimate,使用acquity uplc behc181.7μm (2.1*100mm)色谱柱,自动进样器温度设为8℃,以 0.3ml/min 的流速,50℃的柱温,进样2μl进行梯度洗脱,流动相为:水(0.1%甲酸 10mm甲酸铵)(a)-异丙醇:乙腈=2:5(0.1%甲酸 10mm甲酸铵),梯度洗脱程序为0-5min,70-57%a;5-5.1 min,57-50%a;5.1-14 min,50-30%a;14-14.1min,30%a;14.1-21min,30-1%a;21-24min,1%a;24-24.1min,1-70%a;24.1-28min,70%a。
18.质谱条件:仪器使用thermo q exactive focus,电喷雾离子源(esi),正负离子电离模式,正离子喷雾电压为3.50kv,负离子喷雾电压为2.50kv,鞘气 30arb,辅助气10arb。毛细管温度为325℃,以分辨率35000进行全扫描,扫描范围150-2000,并采用hcd进行二次裂解,碰撞电压为30ev,同时采用动态排除去除无用信息。
19.通过脂质组学进一步确定正常肺泡上皮细胞(hpaepic) 、a549以及akf细胞中哪些脂质发生改变。将hpaepic、a549和akf三株细胞株进行lc-ms和hcd二次裂解后,得到相关脂质代谢数据,脂质代谢物检测结果分析显示,在正常肺泡上皮细胞和肺腺癌细胞之间存在97个差异脂质,其中c16和c18有48个,差异脂质主要类别为甘油三脂(tg)、磷脂酰胆碱(pc)和磷脂酰乙醇胺(pe); akf和野生型a549之间存在102个差异脂质,其中c16和c18的脂质有51个,差异脂质主要类别为tg、pc和pe;综合分析结果见图3-6,图3展示了三种细胞c16和c18差异脂质统计分析热图,图4和图5则为三种细胞c16和c18差异脂质的柱状图汇总;图6则为两组样品中c16和c18差异脂质的韦恩图,从图中可知共有27个相同脂质;结果表明a549细胞敲除far1基因后,a549细胞中部分pc和tg脂质含量可以恢复到正常肺泡细胞的水平,但是会导致a549细胞中部分pe含量的降低。
20.实施例4:qpcr实验分析参与pc、tg合成与代谢的蛋白转录水平的变化为验证差异脂质代谢途径的调控,我们将hpaepic、a549、akf三株细胞提取rna并进行反转录,反转录使用takara反转录试剂盒,并用定量pcr (qpcr)检测参与pc或tg合成与代谢的蛋白的转录情况,pcr扩增引物见表5;表6:引物序列
结果见图7和8,敲除far1基因的a549细胞,参与pc和tg合成与代谢途径蛋白的mrna可以得到恢复,表明far1蛋白参与调控a549细胞的脂质代谢途径。
21.实施例5:敲除far1基因的细胞增殖实验将状态良好,细胞密度为80%左右的细胞(akf、a549、rescue)的培养基去除,用l
×
pbs清洗细胞一次,加适量胰酶,在37℃培养箱中静置3min左右,轻轻拍打培养皿,若较多细胞已被消化离壁,则加入适量的完全培养基终止消化。
22.离心去除上清液,加入适量的培养基稀释细胞沉淀,轻轻的吹打均匀后用血球计数板对三株细胞计数;利用水汽吸附左右将配套的盖玻片紧紧的吸附在计数板凸起的磨砂玻璃条上,使其不能随意移动。吸取10μl细胞悬浮液,从两侧的缝隙中加入到计数板上,由于虹吸原理,细胞悬浮液会均匀的覆盖到计数板和盖玻片中间;对计数板四个角上的4
×
4的方格内的细胞数量进行统计,最后总数再除以4,则是1000μl细胞悬浮液中总的细胞量。(计数后,细胞总数最好在30~100个细胞间以保证计数准确)。
23.将细胞接种于96孔板中,保证每个孔每100μl培养基含有2000个细胞,将细胞置于细胞培养箱中继续培养,细胞接种24h后,取其中6个孔作标记为第0天检测细胞数量,去除培养基,加入含有10% cck8的完全培养基,在细胞培养箱中静置1.5~2h后,用酶标仪检测检测630nm和450nm处的吸光值,此后每天同一时间用同样的操作检测细胞数量,最终数据使用graphpad prism 8软件进行统计分析;结果如图9所示,与野生型a549细胞相比,far1基因的敲除能够抑制a549细胞的增殖。
24.实施例6: 细胞克隆形成实验将状态良好,细胞密度为 80%左右的细胞(akf、a549、rescue)的培养基去除,用1
×
pbs清洗细胞一次,加适量胰酶,在37℃培养箱中静置3min左右,轻轻拍打培养皿,若较多细胞已被消化离壁,则加入适量的完全培养基终止消化。
25.离心去除上清液,加入适量的培养基稀释细胞沉淀,轻轻的吹打均匀后用血球计数板对三株细胞计数。将细胞接种于6孔板中,保证每孔1.5ml完全培养基含有1000个细胞。将细胞置于细胞培养箱中继续培养,每隔4天补充200μl完全培养基。
26.待单个克隆含有50~100个细胞时,去除培养基,用1
×
pbs清洗细胞两次,加入浓
度4%的多聚甲醛固定细胞克隆,待30min后,去除多聚甲醛并用l
×
pbs清洗细胞,加入0.5%的结晶紫染色15min。去除结晶紫,用1
×
pbs清洗细胞2~3次,确保将背景颜色去除;对每孔中的克隆数进行统计,用graphpad prism 8软件进行数据分析;结果见图10,与野生型a549细胞相比,敲除afr1基因后显著抑制a549细胞的克隆形成;表明far1蛋白能够促进a549细胞的克隆形成能力。
27.实施例7: 细胞划痕实验将状态良好,细胞密度为80%左右的细胞(akf、a549、rescue)培养基去除,用1
×
pbs清洗细胞一次,加适量胰酶,在37℃培养箱中静置3min左右,轻轻拍打培养皿,若较多细胞已被消化离壁,则加入适量的完全培养基终止消化。
28.离心去除上清液,加入适量的培养基稀释细胞沉淀,轻轻的吹打均匀后用血球计数板对三株细胞计数;在6孔板中放入伤口愈合插件,使其黏附在6孔板上,每孔先加入100μl 1
×
pbs,轻轻的吹打液体,使每个小室的地步都被浸润到,去除pbs,在每个小室中接种细胞,保证200μl培养基内含有20000个细胞。在插件外围加入1ml培养基,将细胞置于细胞培养箱中继续培养24h后,用镊子小心的将愈合插件拔除,去除培养基后,用1
×
pbs轻轻的清洗细胞,去除未能贴壁的细胞。加入含有20μg/ml的丝裂霉素c的培养基继续培养2小时后换为正常的完全培养基;在10
×
显微镜下拍照,标记为0h,并在24h、48h后在同样为位置拍照并记录;获得的图片用image j软件进行处理后用graphpad prism 8软件进行数据分析。
29.结果见图11,敲除far1基因的a549细胞的迁移能力显著低于野生型a549细胞株,表明far1蛋白能够促进a549细胞的迁移能力。
30.实施例8: 细胞transwell迁移实验将状态良好,细胞密度为80%左右的细胞(akf、a549、rescue)提前饥饿6h,用胰酶将其消化与壁分离进行细胞计数,具体方法如前所述(注:此处重悬细胞时,要用无血清的培养基)。取transwell小室置于24孔板中,每个小室加入100μl含有10000个细胞的悬浮液(不含血清)。沿着24孔板的内壁,缓缓加入500μl完全培养基,将24孔板放入细胞培养箱中继续培养。置于transwell小室中的细胞在渗透压一致的情况下会向营养更丰富的方向迁移。36h后取出transwell小室,轻轻去除小室和24孔板内的培养基,在小室和24孔板中加入适量的4%多聚甲醛固定(确保小室上的细胞可以完全浸润在4%多聚甲醛中),30min后去除多聚甲醛,并用1
×
pbs小心的清洗小室底部,倒置小室控干上面残留水分。待水分控干,在24孔板和transwell小室中加入适量的0.5%结晶紫(确保小室上的细胞可以完全浸润在结晶紫中)进行染色,15min后取出小室,用1
×
pbs小心的对小室进行清洗,直到将背景色洗去,倒置小室,在室温控干小室上残留的水分,控干后,用棉签轻轻的擦拭掉小室内残余的细胞(此处为未迁移的细胞〉,迁移成功的细胞则会保留在小室外侧。用10
×
显微镜对小室拍照,获得的图片用image j软件进行处理后用graphpad prism 8软件进行数据分析。
31.结果见图12,与野生型a549细胞相比,敲除far1基因后细胞迁移能力显著降低,表明far1蛋白能够促进a549细胞的迁移能力。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。