memo1基因的新用途
技术领域
1.本发明属于生物技术领域,具体涉及以memo1基因高表达为目的的筛选用于治疗非小细胞肺癌药物的新用途。
背景技术:
2.肺癌属于世界上死亡率和发病率最高的癌症,肺癌分为小细胞肺癌(small cell lung cancer, sclc)和非小细胞肺癌(non-small cell lung cancer, nsclc)。即使其治疗方法有很多,但在众多的治疗方法中分子靶向治疗具有独特的优势,在肺癌的治疗方案中发挥了十分重要的作用,具有重大的意义。然而,肺癌确诊后的5年存活率为15.6%,低于前列腺癌、乳腺癌或结肠癌的存活率,非小细胞肺癌患者的临床治疗效果并未发生明显改善。因此,寻找更好的非小细胞肺癌临床治疗方案并理解其中的作用机制至关重要。
3.memo1蛋白是erbb2(human epidermal factor receptor 2,人表皮因子受体2)驱动的细胞运动介质1,别称有丙型肝炎病毒ns5a转活蛋白7或c21orf19样蛋白,简称memo1。memo1基因总共包含15个外显子,基因位于智人的2号染色体上,具体位置是2p22.3的31865060bp-32011230bp。memo1蛋白是由297个氨基酸组成,到目前为止memo1蛋白的三维结构已被确定。有研究表明在乳腺癌与结肠癌患者中,memo1蛋白表达水平的增加与患者的不良预后相关,并会降低患者的生存率。memo1蛋白过表达促进癌细胞的迁移、增殖和侵袭,参与癌细胞的转移,抑制细胞凋亡,调节癌细胞的恶性行为。memo1基因在乳腺癌中的机制研究相较而言已经比较深入,但是memo1基因对非小细胞肺癌的具体作用及其作用机制,目前还没有相关文献对这部分进行相应的阐释。
技术实现要素:
4.本发明提供了memo1基因的新用途,即以memo1基因高表达为目的的筛选用于治疗非小细胞肺癌药物的应用,本发明中所述memo1基因的核苷酸序列见ncbi中gene id 51072。
5.为实现上述目的,本发明提供如下技术方案:1、memo1基因敲除质粒px459-ko-memo1的构建在zhang lab(https://zlab.bio/guide-design-resources)网站上选择chopchop,在所有的sgrna中经过多方面综合考虑后选择最佳的一对sgrna进行合成;利用pcr技术将sgrna进行磷酸化和退火,随后连接在px459质粒上,水解线性dna后得到pcr产物即为连接了sgrna的px459-ko-memo1质粒;质粒经转化、挑单个菌落、质粒抽提后利用琼脂糖凝胶电泳和紫外分光光度计检测质粒浓度与纯度,并将与目的片段对应的胶块切下来送到生物公司进行测序;通过生物公司测序结果的序列与sgrna序列进行比对,发现测序结果的px459-ko-memo1质粒上的sgrna与sgrna序列完全一致,即px459-ko-memo1敲除质粒构建成功。
6.2、a549-ko-memo1敲除细胞系的构建
在a549细胞中稳定转染px459-ko-memo1质粒,利用嘌呤霉素筛选出敲除memo1基因的a549稳定细胞系;1
×
sds裂解液(加入β巯基乙醇)提取细胞中的总蛋白,利用蛋白免疫印迹法(western bloting)实验,在蛋白水平验证a549中敲除memo1基因细胞系是否构建成功;进入ncbi找寻memo1基因的cds区,选择合适的碱基序列作为扩增引物,并对引物进行blast后送生物公司合成;提取疑似敲除成功的细胞的全基因组,利用pcr技术将得到的pcr产物进行琼脂糖凝胶电泳,并将与目的片段对应的胶块切下来送到生物公司进行测序,在mrna水平验证a549中敲除memo1基因细胞系是否构建成功;通过dnaman分析软件将测序结果中敲除细胞系的序列与野生型细胞系的序列进行比对,发现敲除细胞系的序列发生了大量的碱基突变,由于这些变化便导致了翻译后memo1基因被成功地敲除;即a459-ko-memo1(akm)敲除细胞系的构建成功。
7.3、检测memo1基因对kegg信号通路富集的影响将野生型a549细胞与敲除型akm细胞进行rna的提取,并分别使用琼脂糖凝胶电泳和agilent 2100进行rna样品的浓度检测,纯度与完整性检测;对检测合格的rna样品进行实验后得到mrna,随后对mrna进行分离和随机打断;并以此为模板合成cdna的第一链和第二链,随后进行cdna的双链纯化;再对纯化后的双链进行末端修复,在片段的3’加入“a”碱基并连接特定的测序接头,最后得到cdna文库;随后对转录组测序文库进行质量评估,使用高通量测序illumina hiseq将满足条件的样品进行上机测序;将原始的测序序列,进行过滤,利用hisat2将过滤得到的clean reads比对到参考基因组序列;基于所选参考基因组序列,使用stringtie软件对mapped reads进行转录本重构,通过分析之后发掘该物种的新转录本和新基因;基于比对结果,将新基因和目前已知基因进行定量分析,对不同样品组中不同基因表达量进行差异基因的表达分析,随后进行差异表达基因的kegg信号通路富集分析,进而检测memo1基因对kegg信号通路富集的影响。
8.4、检测memo1基因对非小细胞肺癌增殖能力的影响将野生型a549细胞与敲除型akm细胞接种至96孔板中,待细胞贴壁后所有孔中都加入pi3k抑制剂(pi3ki:zstk474)进行处理,收集连续四天细胞的od630值与od450值的差值进行实验并分析细胞的增殖能力,通过检测细胞的od值检测memo1基因对非小细胞肺癌增殖能力的影响。
9.将野生型a549细胞与敲除型akm细胞接种至12孔板中,每孔500个细胞,加入1ml完全培养基(高糖dmem培养基,含10%胎牛血清和1%青链霉素)混匀后放入培养箱进行培养;每隔三天对细胞的生长状态进行观察,并进行换液;待每个克隆含有50个细胞左右时对细胞进行固定、染色;通过检测细胞克隆形成率检测memo1基因对非小细胞肺癌增殖能力的影响。
10.5、检测memo1基因对非小细胞肺癌迁移能力的影响将野生型a549细胞与敲除型akm细胞进行6小时的饥饿处理;在transwell小室的上室中加入100μl含有10000个细胞的重悬液,小室内使用无血清培养基;沿着24孔板的内壁加入550μl的完全培养基,使transwell小室与完全培养基液面完全接触;36小时后,取出transwell小室,轻轻去除小室内的无血清培养基,对细胞进行固定、染色;在显微镜下随机取3-5个视野,对小室外侧细胞进行拍照;
提前一天将划痕插件铺入6孔板,将野生型a549细胞与敲除型akm细胞进行正常培养;在插件的每个小格中加入200μl含有30000个细胞的重悬液,在6孔板中划痕插件外加入1ml的完全培养基,细胞培养过夜;取出6孔板,用灭菌镊子将划痕插件轻轻地拔出,吸出完全培养基,磷酸缓冲盐溶液将少量悬浮细胞冲洗掉,加入含有20μg/ml的丝裂霉素c的完全培养基继续培养2小时,再用完全培养基进行换液;在显微镜下进行划痕的拍照,记为0小时;分别在24小时和48小时时间点用同样方法进行拍照。
11.6、检测memo1基因对非小细胞肺癌侵袭能力的影响将野生型a549细胞与敲除型akm细胞进行6小时的饥饿处理;提前用无血清培养基与基质胶按照体积比1:3的比例配制好放在4℃冰箱;加入50μl稀释好的基质胶到小室的上室中并放在37℃培养箱中放置2小时等待胶凝,随后进行基底膜水化;在小室中加入100μl含有10000个细胞的重悬液,小室内使用无血清培养基;沿着24孔板的内壁加入550μl的完全培养基,使transwell小室与完全培养基液面完全接触;36小时后,取出transwell小室,轻轻去除小室内的完全培养基,对细胞进行固定、染色;在显微镜下随机取3-5个视野,对小室外侧细胞进行拍照。
12.7、检测memo1基因对非小细胞肺癌干性能力的影响用成球专用dmem/f12培养基将野生型a549细胞、敲除型akm细胞沉淀重悬起来,dmem/f12培养基中加入20ng/ml的egf、10ng/ml的bfgf以及2%的b27补充剂;在12孔板的每个孔中加入5000个细胞,每种细胞设置3个复孔,混匀后放入细胞培养箱中正常培养;观察细胞成球情况,并进行补液,待细胞球体直径大于50μm时结束培养,拍照并统计球体直径大于50μm的悬浮细胞球数量。
13.8、检测memo1基因对非小细胞肺癌粘附能力的影响用野生型a549细胞与敲除型akm细胞进行细胞粘附实验;根据实验要求分别用10μg/ml牛纤维蛋白原、10μg/ml鼠尾胶原蛋白i和多聚赖氨酸在37℃环境中包被96孔板30分钟,使基质胶聚合成凝胶,随后进行基底膜水化;在96孔板的每孔中加入100μl含10000个细胞的细胞悬液,放入培养箱2小时;除去贴壁不牢的细胞,每孔加入完全培养基100μl,再加入10μl cck-8试剂;培养箱中孵育1小时后用酶标仪进行吸光度值的测定。
14.9、检测memo1基因对非小细胞肺癌药物敏感性的影响将野生型a549细胞与敲除型akm细胞进行化疗药物敏感性实验;按照每孔100μl完全培养基中含有5000个细胞的体系将计数完成的细胞均匀接种至96孔板中,每个实验组至少要有3个平行复孔,将铺好细胞的96孔板放入细胞培养箱中正常培养;待铺板24小时左右细胞完全贴壁后,从细胞培养箱中取出96孔板吸除完全培养基,依次加入100μl含有相应浓度化疗药物的完全培养基;作用于细胞周期的非特异性药物为抑制细胞dna合成的顺铂,主要作用于细胞周期s期,抑制细胞dna合成的5-氟尿嘧啶,对拓扑异构酶ii产生抑制作用的多柔比星,通过抑制拓扑异构酶ii,阻碍dna修复的依托泊苷;5-氟尿嘧啶和顺铂的药物处理时间为36小时;多柔比星的药物处理时间为48小时;依托泊苷的药物处理时间为24小时;药物处理时间到达后,每孔加入10μl cck-8试剂并继续培养1小时;使用酶标仪测定od450值和od630值。
15.10、检测memo1基因对pi3k/akt信号通路的影响使用强ripa裂解液(含1%蛋白酶抑制剂)提取野生型a549细胞与敲除型akm细胞的
总蛋白,并进行定量分析;用pakt、akt、gapdh的一抗进行western bloting实验,验证细胞蛋白水平上pakt与akt的表达状况,检测memo1基因对pi3k/akt信号通路的影响。
16.11、检测memo1基因对slfn11的的影响将野生型a549细胞与敲除型akm细胞用zstk474进行处理后提取总蛋白并进行定量分析;用slfn11、gapdh蛋白的一抗进行western bloting实验,验证细胞蛋白水平上slfn11蛋白的表达状况,检测memo1基因对slfn11的的影响;提取野生型a549细胞与敲除型akm细胞中的mrna,再经过pcr逆转录为cdna,再以此cdna作为qrt-pcr的模板进行qrt-pcr技术,设计slfn11基因的qrt-pcr特异性引物,进而验证细胞mrna水平中slfn11基因的表达状况,并将gapdh作为内参来计算分析实验结果,检测memo1基因对slfn11的的影响。
17.12、检测memo1基因对irs4信号通路的影响提取野生型a549细胞与敲除型akm细胞的总蛋白,用irs4、tublin、isr与gapdh的一抗进行western bloting实验,验证细胞蛋白水平上irs4、tublin、isr与gapdh的表达状况,检测memo1基因对irs4信号通路的影响。
18.本发明的优点和技术效果:本发明实验结果显示a549中敲除memo1基因会对pi3k/akt信号通路的表达产生显著影响、会促进单个细胞和细胞总体的增殖能力,同时对细胞的迁移、侵袭和干性也具有促进作用、会降低细胞的粘附能力、会提高细胞对顺铂、5-fu和依托泊苷的敏感性、会增加细胞中slfn11转录后翻译的表达,并且slfn11蛋白量的变化可以被pi3k的抑制剂zstk474负调控、会增加细胞中胰岛素受体(isr)的表达,并同时增加胰岛素受体底物4(irs4)的表达;即memo1蛋白会抑制a549细胞中slfn11转录后翻译的表达,随后激活pi3k/akt通路,进而在细胞增殖、迁移、侵袭、粘附、干性与药物敏感性等多方面影响肺癌的发生发展进程,如图1所示。
19.实验结果表明促进memo1基因表达的试剂可以用于制备治疗非小细胞肺癌的药物,本发明为非小细胞肺癌药物的治疗提供了一条新的途径。
附图说明
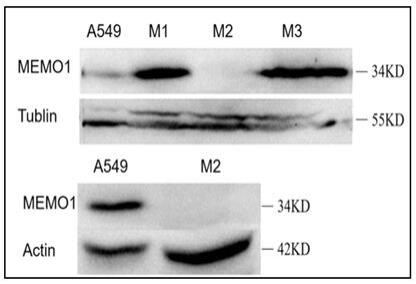
20.图1为本发明中memo1基因触发pi3k/akt的信号通路示意图;图2为利用western bloting检验野生型a549细胞与敲除型akm细胞中memo1蛋白的表达状况;图3为利用琼脂糖凝胶检测野生型a549细胞与敲除型akm细胞全基因组中memo1基因扩增产物的表达结果示意图;图4为野生型a549细胞与敲除型akm细胞全基因组中memo1基因扩增产物的测序结果;图5为本发明中检测memo1基因对kegg信号通路富集的影响结果示意图;图6为利用cck-8实验检验野生型a549细胞与敲除型akm细胞的增殖情况结果;图7为利用cck-8实验检验野生型a549细胞与敲除型akm细胞经过不同zstk474浓度处理后的增殖情况结果;图8为利用克隆形成实验检验野生型a549细胞与敲除型akm细胞的增殖情况(下
图)及其量化图(上图);图9为memo1基因对非小细胞肺癌迁移能力的影响检测结果,其中a图为利用transwell的迁移实验检验野生型a549细胞与敲除型akm细胞的迁移情况(左)及其量化图(右);b图为利用划痕实验检验野生型a549细胞与敲除型akm细胞的迁移情况(左)及其量化图(右);图10为检测memo1基因对非小细胞肺癌侵袭能力影响的结果,其中左中图为拍照结果,右图为统计结果;图11为检测memo1基因对非小细胞肺癌干性能力影响结果;其中:a图为利用成球实验检验a549与akm成球能力的大小(左中图)及其量化图(右图);b图为利用western bloting检验野生型a549细胞与敲除型akm细胞中cd44蛋白的表达状况(左图)及其量化图(右图);图12为检测memo1基因对非小细胞肺癌粘附能力的影响;其中左图使用的基质胶是多聚赖氨酸,中图为使用的基质胶是牛纤维蛋白原,右图使用的基质胶是鼠尾胶原蛋白i;图13为利用药物敏感性实验检验野生型a549细胞与敲除型akm细胞经过不同浓度的顺铂处理后细胞的成活情况(a图)及其分别的ic
50
图(b图);图14为利用药物敏感性实验检验野生型a549细胞与敲除型akm细胞经过不同浓度的5-氟尿嘧啶处理后细胞的成活情况(a图)及其分别的ic
50
图(b图);图15为利用药物敏感性实验检验野生型a549细胞与敲除型akm细胞经过不同浓度的多柔比星处理后细胞的成活情况(a图)及其分别的ic
50
图(b图);图16为利用药物敏感性实验检验野生型a549细胞与敲除型akm细胞经过不同浓度的依托泊苷处理后细胞的成活情况(a图)及其分别的ic
50
图(b图);图17为检测memo1基因对pi3k/akt信号通路的影响的实验结果示意图;图18为检测memo1基因对slfn11的影响的实验结果示意图;其中a为利用western bloting检验野生型a549细胞与敲除型akm细胞中slfn11蛋白的表达状况;b为利用western bloting检验分别经pi3ki与meki处理后野生型a549细胞与敲除型akm细胞中slfn11蛋白的表达状况;c为a图的量化图;d为b图的量化图;图19为利用qrt-pcr实验检验野生型a549细胞与敲除型akm细胞中slfn11基因的mrna水平表达情况;图20为检测memo1基因对irs4信号通路的影响的实验结果示意图;其中a为利用western bloting检验野生型a549细胞与敲除型akm细胞中irs4的蛋白水平表达状况;b为利用western bloting检验野生型a549细胞与敲除型akm细胞中isr的蛋白水平表达状况。
具体实施方式
21.下面结合附图和实施例对本发明作进一步详细说明,但本发明保护范围不局限于所述内容,实施例中使用的试剂和方法,如无特殊说明,均采用常规试剂和使用常规方法;实施例1:memo1基因敲除质粒px459-ko-memo1的构建1、设计与合成sgrna网上搜索zhang lab(https://zlab.bio/guide-design-resources)进入该网站,
tools for guide design选择chopchop,填写目标基因memo1,类别为homo sapiens(hg38/grch38),使用crispr/cas9进行knock-out,再点击find target sites进入下一步,在所有的sgrna中根据里面的排名、效率、所在的外显子数、gc含量、自我互补以及脱靶率等多方面综合考虑后选择一对sgrna(memo1-sgrna-f:5
’‑
caccgtgcattcagctgcggtcctg-3’,memo1-sgrna-r:5
’‑
aaaccaggaccgcagctgaatgcac-3’),送生物公司进行合成。
22.2、sgrna磷酸化和退火根据表1配制磷酸化及退火反应体系;将装有该反应体系的pcr管转移至pcr仪中,利用37℃,30分钟;95℃,5分钟的程序对sgrna进行磷酸化和退火;然后以5℃/min的速度降温至25℃;表1:sgrna磷酸化及退火反应体系3、sgrna与质粒连接按照体积比步骤2产物:无rna酶水=1:199的比例稀释磷酸化的sgrna,稀释后摇匀;根据表2配制sgrna与质粒连接的反应体系,将装有该连接体系的pcr管转移至pcr仪中,按照37℃,5分钟;21℃,5分钟的程序进行六次循环反应;表2 sgrna与质粒连接体系;然后水解未连接成功的线性dna,水解体系如表3,将装有该反应体系的pcr管转移至pcr仪中,按照37℃,30分钟;70℃,30分钟的条件进行pcr;得到的pcr产物即为成功在px459质粒连接了sgrna的px459-ko-memo1质粒;表3水解线性dna体系
;4、px459-ko-memo1质粒转化感受态e.coli从-80℃低温冰箱取出感受态e.coli置于冰上,待用;取步骤3水解产物2μl与50μl感受态e.coli至于ep管中;轻弹ep管底部使水解产物与感受态混匀,冰上放置30分钟;42℃水浴中热激90秒;冰上放置5分钟;加入800μl lb液体培养基,震荡培养50分钟(250r/min,37℃);从摇床上取下菌液ep管,离心(6000r/min,3分钟),在无菌操作台上,吸弃700μl上清液,余下100μl上清液与菌体吹打摇匀;将菌液均匀涂布在amp-lb固体培养基表面;接种完成并等待平板表面干燥后,将amp-lb固体培养基倒置在37℃培养箱中培养13.5小时;同时以转化入px459质粒的菌液为对照;在超净工作台中,按体积比1:1000的比例将浓度100mg/ml 氨苄青霉素(amp)加入lb液体培养基中获得amp-lb液体培养基;选择饱满、单独、不可重叠的菌落,用10μl枪头挑取单克隆菌落至amp-lb液体培养基中(含px459质粒的菌落与含px459-ko-memo1质粒的菌落各挑取3个);在250r/min,37℃的恒温震荡摇床中培养5h;各自取600μl菌液送生物测序;根据测序结果将连接不成功的菌液丢弃,并选择连接成功的菌液进行下一步实验。
23.5、px459-ko-memo1质粒的提取在超净工作台中配制7.5μl amp(100mg/ml) 7.5μl px459-ko-memo1质粒菌液 7.5ml lb液体培养基在50ml离心管中,混匀,同样的操作配制含px459质粒的菌液;将离心管盖子稍微拧松后,将其转移到37℃恒温震荡摇床中,250r/min,培养14.5h;按照碧云天的无内毒素质粒小提中量试剂盒说明书进行质粒提取,提取产物通过琼脂糖凝胶电泳进行检测,并将目的片段对应的胶块送生物公司测序;测序结果表明px459-ko-memo1质粒上的sgrna与sgrna序列完全一致,即px459-ko-memo1质粒构建成功。
24.实施例2:a549-ko-memo1敲除细胞系的构建1、在a549细胞中稳定转染px459-ko-memo1质粒提前六天将培养a549细胞的完全培养基换成无抗生素培养基(高糖dmem培养基,含10%胎牛血清);在六孔板中铺上细胞密度约60%的a549细胞;待细胞贴壁24h后吸弃培养基,用磷酸缓冲盐溶液对细胞进行清洗,随后吸弃磷酸缓冲盐溶液,每孔加入1.5ml optim培养基对a549细胞进行1小时的饥饿处理;在1号ep管中加入200μl optim培养基与2.0μl lipo2000转染试剂,混匀后静置5分钟;在2号ep管中加入200μl optim培养基与1.0μg px459-ko-memo1质粒,混匀后静置5分钟;将上述两个ep管中的溶液混和均匀得到溶液1;将溶液1放置20分钟后直接加入已经进行饥饿处理的a549细胞中,得到转染细胞;将转染细胞放入培养箱中培养5.5小时后换成无
抗生素培养基,继续培养;转染后的第二天,将转染细胞消化下来,扩培到10cm培养皿中继续培养。
25.2、筛选敲除memo1基因的a549细胞系转染后的第三天,更换无抗生素培养基为含有0.8μg/ml嘌呤霉素的嘌呤霉素完全培养基进行阳性克隆细胞的筛选;培养五天更换一次含有0.8μg/ml嘌呤霉素的嘌呤霉素完全培养基,进行4次上述的换液后转染细胞长出单克隆细胞团块;用10μl的移液枪挑取单克隆细胞团块到24孔板中使用完全培养基进行培养;每隔4天进行一次换液,转染细胞培养20天后,扩培到12孔板中继续培养;待转染细胞密度预计为80%时,消化转染细胞,以a549细胞作为对照组细胞;3、蛋白水平验证a549中敲除memo1基因细胞系是否构建成功提取上述转染细胞和a549细胞的蛋白,将细胞沉淀放在4℃冰上;每管加入50μl的1
×
sds裂解液(加入β巯基乙醇),冰上裂解30分钟,每隔10分钟在涡旋震荡仪上震荡一次;98℃金属浴煮样7分钟,待样品冷却后-20℃保存备用;制胶用的玻璃板清洗后烘干,用灭菌水进行检漏,滤纸吸干净,备用;配制浓度为10%的sds聚丙烯酰胺分离胶、浓度为5%的sds聚丙烯酰胺浓缩胶;将提取的蛋白上样后进行80v,35分钟;120v,80分钟的电泳;将预先配置好的转膜液放入4℃预冷,转膜采用三明治-湿转方法,电流290ma转膜1.5h后,用5%脱脂牛奶常温封闭1小时;把配制好的内参与memo1蛋白的一抗与pvdf膜一起4℃摇床孵育过夜,使用1
×
tbst溶液漂洗pvdf膜7分钟,重复3次;加入二抗室温孵育1小时,使用1
×
tbst溶液漂洗pvdf膜8分钟,重复3次;在化学发光仪中进行显影;结果如图2所示,转染细胞m2疑似敲除成功。
26.4、mrna水平验证a549中敲除memo1基因细胞系是否构建成功收集a549细胞与步骤3转染细胞m2的沉淀进行细胞全基因组的提取,提取步骤按照擎科公司的动物基因组dna提取试剂盒的说明书中的方法;在ncbi中找寻memo1基因的cds区并根据sgrna序列所在位置设计扩增引物(memo1-verify-f:5
’‑
atgtccaaccgagtggtctgccgag-3’,memo1-verify-r:5
’‑
cgaaggtcatacagaggtgtcctat-3’),引物进行blast后送生物公司合成;根据表4配制rcr反应体系,按照表5的pcr条件进行目的片段扩增;扩增后的pcr产物进行琼脂糖凝胶,电泳结果见图3所示;将与目的片段大小对应的胶块切下来,送生物公司测序。将测序结果中疑似敲除细胞系的序列与野生型细胞系的序列进行比对,结果如图4所示,结果发现敲除细胞系的序列发生了大量的碱基突变,这些变化便导致了翻译后memo1基因被成功地敲除,即a459-ko-memo1敲除细胞系(akm)构建成功;表4 rcr体系
表5 pcr扩增条件。
27.实施例3:检测memo1基因对kegg信号通路富集的影响提取野生型a549细胞与敲除型akm细胞的rna,并分别使用琼脂糖凝胶电泳和agilent 2100进行rna样品的浓度检测,纯度与完整性检测;对检测合格的rna样品进行实验得到mrna,随后对mrna进行分离和随机打断;并以此为模板合成cdna的第一链和第二链,随后进行cdna的双链纯化;再对纯化后的双链进行末端修复,在片段的3’加入“a”碱基并连接特定的测序接头,最后得到cdna文库;对转录组测序文库进行质量评估,使用高通量测序illumina hiseq将满足条件的样品进行上机测序;将原始的测序序列,进行过滤,利用hisat2将过滤得到的clean reads比对到参考基因组序列(由北京擎科生物科技有限公司完成);基于所选参考基因组序列,使用stringtie软件对mapped reads进行转录本重构,并与原有的基因组注释信息进行比较,寻找原来未被注释的转录区,发掘该物种的新转录本和新基因,从而补充和完善原有的基因组注释信息;过滤掉编码的肽链过短(少于50个 氨基酸残基)或只包含单个外显子的序列,共发掘2482个新基因,其中差异表达基因836个,上调基因284个,下调基因552个。
28.基于比对结果,将得到的新基因和目前已知的基因进行定量分析,对不同样品组中不同基因表达量进行统计分析以及差异基因的表达分析,随后对前面筛选出的差异表达
基因进行kegg信号通路富集分析,进而检测memo1基因对kegg信号通路富集的影响;结果如图5所示,绝大部分信号通路都是由上调基因富集而成。这些信号通路中相较而言更具有显著性差异的信号通路主要集中pi3k/akt信号通路,memo1基因的缺失对pi3k/akt信号通路的表达产生显著影响;这条通路与非小细胞肺癌中memo1蛋白的缺失、slfn11的高表达及非小细胞肺癌的发生发展进程中细胞增殖、迁移、侵袭、干性以及药物敏感性的增加有关。
29.实施例4:memo1基因对非小细胞肺癌增殖能力的影响1、cck-8实验:将野生型a549细胞与敲除型akm细胞按照每孔2500个细胞接种至96孔板中,每组3个复孔,摇晃混匀后轻放入培养箱;铺板24小时后,使用不同浓度的zstk474(浓度为0、0.1、1、10μm)对细胞进行处理,在第一天的孔中加入10μl浓度为10mg/ml的cck-8试剂,培养箱放置1.5小时;使用酶标仪测定od450值和od630值,此时测定的数值为第一天的测量结果;此后,在每天同样的时间对其他的孔加入cck-8试剂进行测定,测定方法同第一天,共计测定4天;结果如图6、7所示,由图可见a549中敲除memo1基因会增加细胞的增殖能力,但zstk474会逆转a549与akm的增殖,即非小细胞肺癌中memo1的缺失会影响pi3k/akt信号通路,进而影响非小细胞肺癌发生发展进程中细胞的增殖能力。
30.2、细胞克隆形成实验:将野生型a549细胞与敲除型akm细胞按照每孔500个细胞接种至12孔板中,加入1ml完全培养基混匀后放入培养箱进行培养;每隔三天观察细胞的生长状态并进行换液;待单个细胞克隆团块的细胞数量大于50个结束细胞培养,吸弃完全培养基,用磷酸缓冲盐溶液清洗细胞;吸弃磷酸缓冲盐溶液,使用浓度为4%的多聚甲醛固定细胞30分钟;固定完毕后吸弃多聚甲醛,用磷酸缓冲盐溶液清洗细胞,加入0.5%结晶紫染液染色15分钟;吸弃结晶紫染液后,用磷酸缓冲盐溶液清洗12孔板5次;结果如图8所示,由图可见,akm细胞的增殖能力比a549细胞的增殖能力更强,memo1基因的敲除会促进a549细胞增殖能力的提高,其中涉及的机制可能也是非小细胞肺癌中memo1的缺失导致pi3k/akt信号通路的激活,进而影响非小细胞肺癌发生发展进程中细胞的增殖能力。
31.实施例5:memo1基因对非小细胞肺癌迁移能力的影响1、野生型a549细胞与敲除型akm细胞进行transwell的迁移实验,野生型a549细胞与敲除型akm细胞提前6小时饥饿处理;将transwell小室放入24孔板中,在transwell小室的上室中加入100μl含有10000个细胞的重悬液(无血清培养基),沿着24孔板的内壁加入550μl的正常培养基,使transwell小室与培养基液面完全接触;将处理好的24孔板放在培养箱中培养36小时后,取出transwell小室,去除小室内的无血清培养基,用磷酸缓冲盐溶液清洗细胞;吸弃磷酸缓冲盐溶液,使用浓度为4%的多聚甲醛固定细胞30分钟;固定完毕后吸弃多聚甲醛,用磷酸缓冲盐溶液清洗细胞,加入0.5%结晶紫染液染色15分钟;吸弃结晶紫染液后,用清水将小室冲洗至流过小室地液体为无色,在室温将小室吹干;擦除上室中的细胞,随机取3-5个视野,对小室外侧细胞进行拍照;结果如图9a所示,由图可见,akm迁移到下室的细胞数量远多于a549,memo1基因抑制a549细胞的迁移能力。
32.2、野生型a549细胞与敲除型akm细胞进行划痕实验,在进行划痕实验前一天将划
痕插件放入6孔板中;野生型a549细胞与敲除型akm细胞进行正常培养;在划痕插件的每个小格中加入含有30000个细胞的200μl完全培养基;在6孔板的划痕插件外加入1ml完全培养基,细胞培养过夜;用灭菌镊子将划痕插件轻轻地拔出,吸弃完全培养基,磷酸缓冲盐溶液将少量悬浮细胞冲洗掉;加入含有20μg/ml的丝裂霉素c的完全培养基继续培养2小时,随后用完全培养基进行换液;在显微镜下进行划痕的拍照,记为0小时,之后分别在24小时和48小时时间点用同样方法进行拍照;结果如图9b所示,由图可见,a549中memo1基因的敲除会加大愈合百分比,显示出更强的迁移能力,而这个结果也和traswell迁移实验的结果一致,即a549中memo1基因的敲除会促进细胞迁移能力,memo1基因抑制a549细胞的迁移能力。
33.实施例6:memo1基因对非小细胞肺癌侵袭能力的影响采用野生型a549细胞与敲除型akm细胞进行transwell的侵袭实验,野生型a549细胞与敲除型akm细胞提前6小时饥饿处理;将无血清培养基与基质胶按照1:3的比例配制,加入50μl已稀释的基质胶到小室的上室中,并在37℃培养箱中放置2小时;随后加入20μl 磷酸缓冲盐溶液到小室的上室中,在37℃培养箱中放置30分钟;将经上述处理后的transwell小室放入24孔板中,在transwell小室的上室中加入含10000个细胞的无血清培养基100μl,24孔板中加入550μl完全培养基;细胞培养36小时后,去除transwell小室内的无血清培养基,用磷酸缓冲盐溶液清洗细胞;吸弃磷酸缓冲盐溶液,使用浓度为4%的多聚甲醛固定细胞30分钟;固定完毕后吸弃多聚甲醛,用磷酸缓冲盐溶液清洗细胞,加入0.5%结晶紫染液染色15分钟;吸弃结晶紫染液后,用清水将小室冲洗至流过小室地液体为无色,在室温将小室吹干;擦除上室中的细胞,随机取3-5个视野,对小室外侧细胞进行拍照;结果如图10所示,由图可见,akm的细胞数量远多于a549,即a549中memo1基因的敲除会促进细胞的侵袭能力,memo1基因抑制a549细胞的侵袭能力。
34.实施例7:检测memo1基因对非小细胞肺癌干性能力的影响采用野生型a549细胞与敲除型akm细胞进行细胞成球实验,成球实验所用培养基为成球专用dmem/f12培养基,dmem/f12培养基中加入20ng/ml的egf、10ng/ml的bfgf以及2%的b27补充剂;将野生型a549细胞与敲除型akm细胞接种至12孔板中,每孔中5000个细胞,每种细胞设置3个复孔,混匀后放入细胞培养箱中正常培养;每天轻轻晃动12孔板,每隔3天观察细胞并补加50μl上述培养基;细胞培养14天后拍照并统计球体直径大于50μm的悬浮细胞球数量,结果如图11a所示;western bloting实验:对使用完全培养基培养的a549和akm细胞进行总蛋白的提取,利用western bloting检测了cd44蛋白的表达情况,结果如图11b所示;由图11可见,akm细胞的球体个数或者是球体的直径显著地多于对照组;cd44蛋白被认为是干细胞的细胞表面标志物,akm细胞中cd44蛋白的表达量远远高于a549,综上,a549中memo1基因的敲除增加了具有典型癌干细胞样表型的肺癌细胞群体,memo1基因抑制a549细胞的干性能力。
35.实施例7:memo1基因对非小细胞肺癌粘附能力的影响采用野生型a549细胞与敲除型akm细胞进行细胞粘附实验,分别使用10μg/ml牛纤维蛋白原、10μg/ml鼠尾胶原蛋白i和多聚赖氨酸在37℃环境中包被96孔板30分钟;随后在每个孔中加入50μl 磷酸缓冲盐溶液,37℃培养箱中放置30分钟,吸弃多余的磷酸缓冲盐溶
液;在96孔板的每孔中加入100μl含10000个细胞的细胞悬液,37℃培养2小时;吸弃完全培养基,轻轻地用磷酸缓冲盐溶液清洗细胞表面,除去贴壁不牢的细胞;每孔加入完全培养基100μl,再加入10μl cck-8试剂;培养箱中孵育1小时后使用酶标仪进行od值的测定;结果如图12所示,由图可见,akm的贴壁细胞个数与效果都显著性地弱于a549细胞,在a549中敲除memo1基因后会降低细胞的粘附能力,memo1基因促进a549细胞的粘附能力。
36.实施例8:检测memo1基因对非小细胞肺癌药物敏感性的影响采用野生型a549细胞与敲除型akm细胞进行化疗药物敏感性实验,将野生型a549细胞与敲除型akm细胞按照每孔5000个细胞,100μl完全培养基接种至96孔板中,每组3个平行复孔;24小时左右细胞完全贴壁后,吸弃完全培养基,依次加入不同处理时间、不同浓度的不同化疗药物;顺铂的药物浓度为0、1、10、50、100μm,处理时间为36小时;5-氟尿嘧啶的药物浓度为0、1、10、100、1000μg/ml,处理时间为36小时;多柔比星的药物浓度为0、100、500、1000、2000nm,处理时间为48小时;依托泊苷的药物浓度为0、1、10、50、100μm,处理时间为24小时;药物处理时间到达后,每孔加入10μl cck-8试剂并继续培养1小时;使用酶标仪测定od450值和od630值;结果如图13、14、15、16所示,由图可见,akm相较于a549而言,akm在5-氟尿嘧啶、顺铂和依托泊苷的处理下药物敏感性增加、细胞死亡率增加、存活率降低,而且都具有显著性差异,尤其是依托泊苷,但是多柔比星对两种细胞处理后,这两种细胞之间并没有表现出明显的差异。就ic
50
上而言,memo1基因的敲除使得顺铂和5-氟尿嘧啶的ic
50
值分别降低为30μm和1μg/ml,而对于依托泊苷而言,ic
50
之间的差异性特别显著,也就是说a549细胞对于这顺铂、依托泊苷和5-氟尿嘧啶的敏感性与memo1蛋白的表达量呈负相关,而这三种药物都是与dna有密切的关系,总而言之,a549中memo1基因的敲除会增加细胞对顺铂、依托泊苷和5-氟尿嘧啶的敏感性,memo1基因抑制a549细胞对顺铂、依托泊苷和5-氟尿嘧啶的敏感性。
37.实施例9:memo1基因对pi3k/akt信号通路的影响提取野生型a549细胞与敲除型akm细胞的总蛋白,提前一天将野生型a549细胞与敲除型akm细胞铺入六孔板中,培养至细胞密度为80%,吸弃完全培养基,每孔加入120μl强ripa裂解液(含1%蛋白酶抑制剂);在冰上进行30min的细胞裂解,每隔10min将六孔板置于振荡器上震荡1min;将细胞裂解样收集到ep管中,4℃、15000r/min离心15 分钟;将离心完的样品放于冰上,取上清液于提前做好预冷和标记的新ep管中;蛋白定量分析,用移液枪将蛋白标准溶液(0.5μg/μl)吸入96孔板中,随后按浓度梯度(0、0.5、1、2、4、6、8、10μg/μl)将溶液进行稀释,使得每孔中溶液终体积为20μl,体积不足用磷酸盐缓冲液进行补齐;96孔板每孔一一对应加入2μl细胞裂解液样品、18μl磷酸盐缓冲液;所有孔中加入200μl bca工作液(bca工作液由a液与b液按照50:1的比例配制而成),将96孔板放入37℃恒温箱中培养30min;使用酶标仪测定od562值,计算样品蛋白质的质量;之后加入占总体积1/5的5
×
loadin;将样品混匀瞬离,在98℃金属浴中煮样7 min,待样品自然冷却后-20℃保存待用;用pakt、akt、gapdh的一抗进行western bloting实验,验证细胞蛋白水平上pakt与akt的表达状况,检测memo1基因对pi3k/akt信号通路的影响;结果如图17所示,由图可见,a549中敲除memo1基因会降低细胞中akt的表达,增加
akt的磷酸化水平,即memo1基因的缺失会导致pi3k/akt信号通路的激活,进而影响非小细胞肺癌发生发展进程。
38.实施例10:memo1基因对slfn11蛋白的影响野生型a549细胞与敲除型akm细胞用zstk474进行处理后提取总蛋白,并进行蛋白质定量分析(蛋白提取与定量方法同实施例9),用slfn11、gapdh蛋白的一抗进行western bloting实验,验证细胞蛋白水平上slfn11蛋白的表达状况,检测memo1基因对slfn11蛋白的的影响,结果如图18所示;提取野生型a549细胞与敲除型akm细胞中的mrna,经过pcr技术逆转录获得cdna,再以此cdna作为qrt-pcr的模板进行qrt-pcr技术,验证细胞mrna水平中slfn11基因的表达状况,slfn11的qrt-pcr引物为slfn11-qpcr-f:5
’‑
gaaacgctaagggggcttcg-3’,slfn11-qpcr-r:5
’‑
acgggtagaaacgcaactcc-3’,并将gapdh作为内参来计算分析实验结果,检测memo1基因对slfn11的影响,实验结果如图19所示;由图可见,slfn11蛋白在a549中低表达,a549中敲除memo1基因会逆转slfn11蛋白在a549中的低表达,并且对slfn11基因的转录水平无关,即memo1基因影响是slfn11转录后翻译的表达,同时上述蛋白质的变化情况是可以被pi3k的抑制剂zstk474负调控,即memo基因的缺失只在蛋白水平上影响slfn11蛋白的表达,而这一过程中还有pi3k/akt信号通路参与其中,共同影响癌症进程。
39.实施例11:memo1基因对irs4信号通路的影响提取野生型a549细胞与敲除型akm细胞的总蛋白,并进行蛋白质定量分析(蛋白提取与定量方法同实施例9),用irs4、tublin、isr与gapdh的一抗进行western bloting实验,验证细胞蛋白水平上irs4、tublin、isr与gapdh的表达状况,检测memo1基因对irs4信号通路的影响;结果如图20所示,由图可见,a549中敲除memo1基因会增加细胞中胰岛素受体的表达,并同时增加胰岛素受体底物4的表达,即memo1基因也影响irs4信号通路,在非小细胞肺癌中参与的不是简单的一条通路,而是通过多条通路共同调控整个过程。
40.本发明利用crispr/cas9技术在非小细胞肺癌a549细胞中敲除了memo1基因,通过生物信息学分析与实验检测发现a549中敲除memo1基因基因会对pi3k/akt信号通路的表达产生显著影响、会促进单个细胞和细胞总体的增殖能力,同时对细胞的迁移、侵袭和干性也具有促进作用、会降低细胞的粘附能力、会提高细胞对顺铂、5-氟尿嘧啶和依托泊苷的敏感性、会增加细胞中slfn11转录后翻译的表达,并且slfn11蛋白的变化可以被pi3k的抑制剂zstk474负调控、会增加细胞中胰岛素受体的表达,并同时增加胰岛素受体底物4的表达。即memo1蛋白会抑制a549细胞中slfn11转录后翻译的表达,随后激活pi3k/akt通路,进而在细胞增殖、迁移、侵袭、粘附、干性与药物敏感性等多方面影响肺癌的发生发展进程。
41.这些研究内容表明可用于促进memo1基因表达的试剂可以用于制备治疗非小细胞肺癌的药物,也表明可用于抑制slfn11基因表达的试剂可以用于制备治疗非小细胞肺癌的药物,本发明为非小细胞肺癌药物的治疗提供了一条新的途径,将有助于本文筛选出更有效的药物治疗新靶点和更多的生物标志物,以便于临床上更好的治疗肺癌,提高肺癌患者的生活质量。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。