一种确定tat细胞的方法
技术领域
1.本发明提供了一种确定血液中的tat细胞的方法,涉及癌症治疗、筛查、检测与评估领域。
背景技术:
2.肿瘤液体活检是当前癌症筛查领域极具前景的技术手段,能够以非侵入的方式采集患者样本,操作简便且对患者的创伤较小。过往基于血清癌症标志物、cell-free dna(cfdna)、circulating tumor cells(ctc)等进行肿瘤液体活检的技术手段,多是从肿瘤相关的特性出发,在肿瘤发展的早期无法被有效地检测到,信噪比很低。机体的免疫系统具有识别肿瘤抗原并将其消灭清除的作用,特别是t淋巴细胞介导的适应性免疫应答,能够迅速激活免疫反应,防止疾病进展。因此理论上t淋巴细胞的免疫应答是早于肿瘤发生的。
3.t淋巴细胞有两类重要的功能特征:第一类是t淋巴细胞表面特异性识别抗原和介导免疫应答的t cell receptor(tcr)受体分子,不同的tcr序列对于抗原信息的识别具有特异性。同一个样本内所有tcr受体序列的集合也称为tcr序列组库,能够反映机体的适应性免疫应答,从而间接表征疾病进程状态;第二类是在tcr序列识别抗原信息后激活细胞内的免疫信号通路,表现在t细胞基因转录组层面的变化。这两类特征相互偶联但又不完全相关,共同发挥适应性免疫应答的肿瘤杀伤功能。
4.由于肿瘤细胞高频的突变效应,在应对机体的免疫选择中会发生免疫逃逸的现象,因此机体中存在大量与肿瘤抗原识别无关的t细胞,也称为旁观者t细胞。现有的富集外周血中肿瘤特异性的t淋巴细胞需要事先构造肿瘤特异性的抗原肽段,然后利用体外刺激实验等策略去筛选能够和抗原肽段相互作用的t淋巴细胞。这样的方法通量低、实验繁琐且没有办法覆盖所有和肿瘤识别相关的t细胞。本发明则在血液中界定出一群肿瘤相关t淋巴细胞(tumor associated t cells,tat)-tat细胞,提高筛选方法的灵敏度。
5.本发明涉及利用外周血t淋巴细胞受体(tcr)序列组库以及转录组两方面特征,构建用于肿瘤筛查的预测模型,完全从免疫应答的视角去进行癌症的液体活检。本发明方法仅需要采集患者外周血标本,侵入性小且可利用的信息多,能对现有的肿瘤液体活检方法学做出非常好的补充,且有潜力检测早期无症状肿瘤,真正将癌症诊疗一体化前置,具有非常重要的应用价值。
技术实现要素:
6.本发明通过分析确定出血液中的一类t细胞,将其命名为tat细胞,该类t细胞具有的tcr也存在于同一受试者的肿瘤浸润的t细胞中。本发明进一步发现这一群tat细胞具有较高的克隆增殖功能,可以反映外周血中肿瘤相关的t细胞群体,同时也预期具有更强的与肿瘤细胞特异性结合的能力。
7.在一个方面,本发明提供了一种tat细胞,其包含tat-tcr序列,所述tat-tcr序列是在血液中的t细胞和肿瘤浸润的t细胞中共同存在的tcr序列;优选地,在一些实施方式
中,tat-tcr序列不是非癌症相关t细胞tcr序列。
8.在一个方面,本发明提供了一种确定tat细胞的方法,其包括判断待确定t细胞的tcr序列是否为tat-tcr序列,由此确定所述待确定t细胞是否为tat细胞。
9.在一些实施方式中,确定tat细胞的方法还包括获得tat-tcr序列集,并且(1)判断待确定t细胞的tcr序列是否存在于所述tat-tcr序列集中,由此判断待确定t细胞的tcr序列是否为tat-tcr序列;或者(2)根据对所述tat-tcr序列集进行计算产生的模型,判断待确定t细胞的tcr序列是否为tat-tcr序列。
10.在一些实施方式中,tat-tcr序列集是预先确定的tat-tcr序列。在一些实施方式中,预先确定是从已公开的数据集中确定的或是对已知患有肿瘤的受试者进行测序并分析获得的。
11.在一些实施方式中,预先确定是从已公开的数据集中获得配对的血液中t细胞的tcr序列和肿瘤浸润的t细胞中的tcr序列,并且将配对的血液中t细胞的tcr序列和肿瘤浸润的t细胞中的tcr序列中共同存在的tcr序列作为预先确定的tat-tcr序列;优选地,在一些实施方式中,所述预先确定的tat-tcr序列不是非癌症相关t细胞tcr序列。
12.在一些实施方式中,所述预先确定是测定已知患有肿瘤的受试者的配对的血液中t细胞的tcr序列和肿瘤浸润的t细胞中的tcr序列,并且将配对的血液中t细胞的tcr序列和肿瘤浸润的t细胞中的tcr序列中共同存在的tcr序列作为预先确定的tat-tcr序列;优选地,在一些实施方式中,所述预先确定的tat-tcr序列不是非癌症相关t细胞tcr序列。
13.在一些实施方式中,所述计算是使用机器学习模型对所述tat-tcr序列集进行计算。
14.在一些实施方式中,机器学习模型是逻辑回归模型、支持向量机模型、卷积神经网络等深度学习框架。优选地,在一些实施方式中,机器学习模型是卷积神经网络。
15.在一个方面,本发明提供了一种评估受试者癌症风险的方法,其包括(1)根据本发明的方法确定所述受试者血液中的tat细胞;(2)确定所述tat细胞在所述受试者血液中的富集程度;根据所述富集程度评估受试者的癌症风险。
16.在一个方面,本发明提供了一种评估受试者癌症风险的方法,其包括(1)测定所述受试者的pbmc细胞中的tat特征基因集的基因表达量;根据所述基因表达量评估受试者的癌症风险;其中所述tat特征基因集是tat细胞相对于非癌症相关t细胞具有差异表达的基因;所述tat特征基因集是预先确定的。
17.在一个方面,本发明提供了一种评估受试者癌症风险的方法,其包括(1)根据本发明的方法确定所述受试者血液中的tat细胞;(2)确定所述tat细胞在所述受试者血液中的富集程度;(3)测定所述受试者的pbmc细胞中的tat特征基因集的基因表达量;根据所述富集程度和所述基因表达量评估受试者的癌症风险;其中所述tat特征基因集是预先确定的。
18.在一些实施方式中,tat特征基因集是预先确定的是指从已公开的数据集中确定的或是对已知患有肿瘤的受试者进行测序并分析确定的。在一些实施方式中,从已公开的数据集中确定是指从已公开的数据集中获得或经分析获得tat细胞的转录组信息和非癌症相关t细胞的转录组信息,比较所述tat细胞的转录组信息和所述非癌症相关t细胞的转录组信息,将经所述比较获得的差异表达的基因作为tat特征基因集。
19.在一些实施方式中,对已知患有肿瘤的受试者进行测序并分析确定是指对已知患
有肿瘤的受试者的t细胞进行测序并根据本发明的方法确定所述受试者的tat细胞和非癌症相关t细胞;比较所述tat细胞的转录组信息和所述非癌症相关t细胞的转录组信息,将经所述比较获得的差异表达的基因作为tat特征基因集。
20.在一些实施方式中,pbmc细胞是t细胞或根据本发明的方法确定的所述受试者的tat细胞。
21.在一个方面,本发明提供了用于检测受试者血液中t细胞的tcr序列的试剂在制备用于评估癌症风险的试剂盒中的用途。
22.在一个方面,本发明提供了用于检测受试者血液中pbmc细胞的tat特征基因集的基因表达量的试剂在制备用于评估癌症风险的试剂盒中的用途。
23.在一个方面,本发明提供了用于检测受试者血液中t细胞的tcr序列的试剂和用于检测受试者血液中pbmc细胞的tat特征基因集的基因表达量的试剂在制备用于评估癌症风险的试剂盒中的用途。
24.在一个方面,本发明提供了用于检测受试者血液中t细胞的tcr序列的试剂,其用于评估受试者的癌症风险。
25.在一个方面,本发明提供了用于检测受试者血液中pbmc细胞的tat特征基因集的基因表达量的试剂,其用于评估受试者的癌症风险。
26.在一些实施方式中,pbmc细胞是t细胞或根据本发明的方法确定的所述受试者的tat细胞。
27.在一些实施方式中,tat-tcr序列是tat细胞的tcr3序列。
附图说明
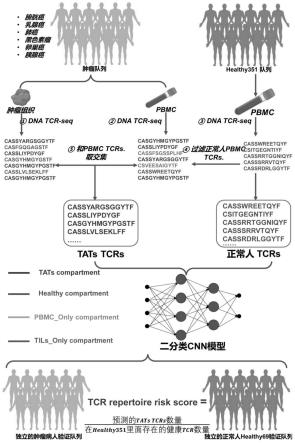
28.图1:确定tat-tcr序列策略以及使用tat-tcr序列预测癌症风险的示意图;
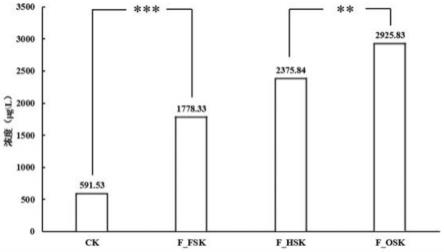
29.图2:多种癌症中的trrs值高于健康人群或病毒感染人群中的trrs值;
30.图3:tat细胞克隆性增殖能力的分析;
31.图4:(a)14个数据集中的tat细胞中的一些肿瘤杀伤,免疫调节等相关基因的表达强度显著高于非癌症相关t细胞中的相关基因表达强度;(b)一个数据集中的tat特征基因能够独立预测其他数据集中的细胞的种类。
32.图5:联合使用tat-tcr序列和tat特征基因集来评估癌症风险的策略;
33.图6:验证联合使用tat-tcr序列和tat特征基因集来评估癌症风险的能力。
具体实施方式
34.在本说明书全文,提到“一个实施方式”、“实施方式”、“特定实施方式”、“相关实施方式”、“某个实施方式”、“另外的实施方式”或“进一步的实施方式”或其组合意指与所述实施方式结合描述的特定特征、结构或特性被包括在本发明的至少一个实施方式中。因此,前述短语在本说明书全文的各个地方的出现不一定全部指相同实施方式。此外,特定特征、结构或特性可以以任何合适方式在一个或多个实施方式中组合。
[0035]“由
……
组成”意指包括但限于在短语“由
……
组成”后的任何。因此,短语“由
……
组成”是指示所列出的要素是需要的或强制性的,并且没有其他要素是可以存在的。
[0036]
概述
[0037]
本发明的方法涉及从外周血中确定出能够反映肿瘤微环境中能够和肿瘤抗原结合能力更强的一群t细胞亚群。
[0038]
除非另有定义,否则本文使用的所有技术和科学术语具有与本发明所属领域普通技术人员通常理解的相同的含义。为了本发明的目的,下文定义了下述术语。
[0039]
本文所述“肿瘤相关t淋巴细胞”(tumor associated t cells,tat)或“tat细胞”是指癌症病人血液中的一种t细胞,该种t细胞具有的tcr序列同时存在于该病人的肿瘤浸润的t细胞中。本领域技术人员能够理解,相对于血液中的其他t细胞,tat细胞结合该病人的肿瘤抗原的能力更强;相对于血液中的其他t细胞,预期tat细胞杀伤肿瘤细胞的能力更强。
[0040]
本文所述“非癌症相关t细胞”包括受试者中tat细胞以外的其他t细胞。在一些实施方式中,所述“非癌症相关t细胞”是指健康受试者体内的t细胞。在一些实施方式中,所述“非癌症相关t细胞”是指健康受试者体内的除tat细胞以外的其他t细胞。
[0041]
本文所述“tat-tcr序列”是指tat细胞中的tcr序列,其包括tcr1、tcr2和tcr3序列。在一些实施方式中,tat-tcr序列是tat细胞中的tcr3序列。在一些实施方式中,tcr序列是tcr3序列。
[0042]
本文所述“癌症风险”是指受试者患有癌症的风险,即受试者患有癌症的可能性。其可以以百分比的形式体现。
[0043]
本文所述“序列集”是指序列的集合,其包含一条或多条序列。
[0044]
本文所述“受试者”,指人或动物,包括所有哺乳动物,比如灵长类(尤其高级灵长类)、绵羊、狗、啮齿动物(例如,小鼠或大鼠)、荷兰猪、山羊、猪、猫、兔子和母牛。本文所述“健康受试者”是指利用现有手段或本领域常规的医学评估方法,没有被认定为患有癌症的受试者。
[0045]
本文所述“已公开的数据集”是指本领域技术人员能够从公开发表的渠道获知的任何数据信息。
[0046]
本文所述“配对的血液中t细胞的tcr序列和肿瘤浸润的t细胞中的tcr序列”是指一对来自同一受试者的血液中的t细胞tcr序列和肿瘤浸润的t细胞的tcr序列或多对。
[0047]
本文所述“富集程度”是指任何能够反映tat细胞在血液中的绝对量或相对量的指标。在一些实施方式中,可以使用tat细胞占血液中所有细胞或占血液中所有t细胞的百分比或两者之间的比例作为该指标。在一些实施方式中,可以使用tat细胞在每毫升血液中的数量作为该指标。
[0048]
本文所述“tat特征基因集”是指相对于非癌症相关t细胞的基因表达,tat细胞中具有差异表达的基因集。本领域技术人员理解,能够使用本领域任何常规的测序技术,对比分析tat细胞和非癌症相关t细胞的差异表达的基因,由此得出tat特征基因集。
[0049]
本文所述“转录组信息”是指细胞中部分或所有(即一个或多个基因)基因表达量(如相对量和/或绝对量)等信息。本领域技术人员能够理解,可以使用本领域任何常规技术(如rna测序等)确定转录组信息。
[0050]
可以使用本领域任何常规技术确定t细胞中的tcr序列,其包括但不限于sanger测序,常用的二代测序、三代测序技术等。
[0051]
可以使用本领域任何常规的技术确定t细胞中的转录组信息或测定t细胞中的基
因表达的量。
[0052]
优选地,在一些实施方式中,可以使用单细胞测序技术确定t细胞群中每个t细胞的tcr序列或转录组信息。
[0053]
tat-tcr序列集的确定
[0054]
发明人发现,癌症患者的血液中存在一种和肿瘤浸润的t细胞具有相同tcr序列的t细胞,并将该种t细胞命名为tat细胞。tat细胞中的tcr序列组成的集合为tat-tcr序列集。该序列集可以用于识别与肿瘤细胞结合能力更强的t细胞。
[0055]
相比于非癌症相关t细胞,包含tat-tcr序列的t细胞在肿瘤抗原刺激下的克隆性增殖能力更强,与肿瘤细胞的结合能力也更强,预期杀伤肿瘤细胞的效果也更强。
[0056]
可以使用本领域任何已知的方法确定tat-tcr序列集。在一些实施方式中,确定血液中tat-tcr序列集的方法包括(1)获得癌症患者血液中的t细胞的tcr序列集-1;(2)获得癌症患者肿瘤浸润的t细胞的tcr序列集-2;(3)获得序列集-1和序列集-2中共同出现的序列集-3;将序列集-3作为tat-tcr序列集。
[0057]
在一些实施方式中,确定血液中tat-tcr序列集的方法包括(1)获得癌症患者血液中的t细胞的tcr序列集-1;(2)获得癌症患者肿瘤浸润的t细胞的tcr序列集-2;(3)获得序列集-1和序列集-2中共同出现的序列集-3;(4)从序列集-3中去除健康受试者中出现的tcr序列集-4得到tcr序列集-5;将所述序列集-5作为tat-tcr序列集。
[0058]
在一些实施方式中,所述序列集-1、序列集-2和序列集-4来自于已公开的数据集。
[0059]
在一些实施方式中,所述序列集-1和序列集-2可以通过对一个或多个癌症患者的t细胞使用常规方法进行tcr测序来获得。
[0060]
在一些实施方式中,序列集-4可以通过对一个或多个健康受试者的t细胞使用常规方法进行tcr测序来获得。
[0061]
在一些实施方式中,序列集-1中的序列数目为1、10、100、1000、10000、100000、1000000条或更多、或上述数值之间的任意值。在一些实施方式中,序列集-2中的序列数目为1、10、100、1000、10000、100000、1000000条或更多、或上述数值之间的任意值。在一些实施方式中,序列集-4中的序列数目为1、10、100、1000、10000、100000、1000000条或更多、或上述数值之间的任意值。
[0062]
tat细胞的确定
[0063]
在一些实施方式中,将机器学习模型应用到序列集-3或序列集-5中进行计算,以产生或训练出能够计算一条给定的tcr序列是tat-tcr序列的概率的模型。
[0064]
在一些实施方式中,将待确定t细胞的tcr序列应用到该模型中计算出其中每条tcr序列是tat-tcr序列的概率。可以设置一个给定的截取值(cut-off),并将高于该截取值的概率的tcr序列确定为是tat-tcr序列,将包含tat-tcr序列的t细胞确定为tat细胞。
[0065]
在一些实施方式中,该截取值是66%、70%、75%或更高或上述数值之间的任意值。
[0066]
在一些实施方式中,上述机器学习模型可以是逻辑回归模型、支持向量机模型、卷积神经网络等深度学习框架。
[0067]
癌症风险的评估方法
[0068]
(1)使用受试者血液中tat细胞的富集程度来分析、预测或评估受试者的癌症风
险。
[0069]
可以使用受试者中血液中的tat细胞的富集程度来分析、预测或评估受试者的癌症风险。发明人发现,该富集程度与受试者的癌症风险呈正相关性,因而能够用于分析、预测或评估受试者的癌症风险。
[0070]
在一些实施方式中,可以使用tcr库风险评分(trrs)来评估癌症风险,所述trrs是利用测得的受试者血液中的tat-tcr序列数目除以受试者血液中的非癌症相关t细胞的tcr序列数目得到的比例。在一些实施方式中,可以将健康受试者中的tcr序列作为非癌症相关t细胞的tcr序列。
[0071]
该trrs能够反映出受试者血液中tat细胞的富集程度,因而也能够用于分析、预测或评估受试者的癌症风险。
[0072]
在一些实施方式中,可以将受试者血液中的tat-tcr序列数目除以受试者血液中的所有t细胞的tcr序列数目得到一个比例。该比例可命名为总风险评分。本领域技术人员能够理解,该总风险评分也与受试者的癌症风险呈正相关性。
[0073]
本领域技术人员能够理解,任何能够反映tat细胞在受试者在血液中的富集程度的指标均与该受试者的癌症风险呈正相关性,因此也均可用于分析、预测或评估受试者的癌症风险。
[0074]
(2)使用受试者外周血单个核细胞(pbmc细胞)中的tat特征基因集的基因表达量来分析、预测或评估受试者的癌症风险。
[0075]
可以使用受试者pbmc细胞的转录组信息来分析、预测或评估受试者的癌症风险。在一些实施方式中,pbmc细胞是t细胞或根据本发明的方法确定的tat细胞。
[0076]
发明人还发现,相对于非癌症相关t细胞,tat细胞中的一些基因能够表达程度显著升高。这些显著升高的差异表达基因主要富集在t细胞增殖、t细胞激活、pd-1信号通路、mhc介导的抗原多肽组装与呈递、淋巴细介导的免疫应答等模块。在一些实施方式中,这些模块可以被当作tat特征基因集。在一些实施方式中,将tat细胞中表达量相对于非癌症相关t细胞具有统计学上的显著性差异的基因作为tat特征基因集中的基因。在一些实施方式中,将tat细胞中表达量相对于非癌症相关t细胞中的差异在5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%或更多或上述数值之间的任意值的基因作为tat特征基因集中的基因。
[0077]
在一些实施方式中,可以使用pbmc细胞中的tat特征基因集的基因表达量来分析、预测或评估受试者的癌症风险。在一些实施方式中,该tat特征基因集的基因表达量可以通过测序的方式获得,优选地使用单细胞测序的方式获得。
[0078]
在一些实施方式中,tat特征基因集是包含t细胞增殖、t细胞激活、pd-1信号通路、mhc介导的抗原多肽组装与呈递或淋巴细介导的免疫应答等模块的基因。
[0079]
本领域技术人员能够理解,任何能够反映tat特征基因集的基因表达量相对于非癌症细胞(如非癌症相关t细胞)中该tat特征基因集的基因表达量的差异的指标,均可用于分析、预测或评估受试者的癌症风险。在一些实施方式中,将tat特征基因集的基因表达量应用到本领域常用的计算基因表达富集特征的模型如genemodulescore(hao y,hao s,andersen-nissen e,et al(2021)integrated analysis of multimodal single-cell data.cell)中,获得tat富集得分,该tat富集得分能够反映,相对于另一细胞群(如非癌症
相关t细胞),tat特征基因集的基因表达量的差异或富集情况。
[0080]
(3)使用受试者血液中的tat细胞的富集程度和受试者外周血单个核细胞(pbmc细胞)中的tat特征基因集的基因表达量来分析、预测或评估受试者的癌症风险。
[0081]
可以使用受试者血液中的tat细胞的富集程度和tat特征基因集的基因表达量来分析、预测或评估受试者的癌症风险。在一些实施方式中,pbmc细胞是t细胞或根据本发明的方法确定的tat细胞。
[0082]
在一些实施方式中,可以使用任何能够反映受试者血液中的tat细胞的富集程度的指标或数值(如本文记载的)与pbmc细胞的tat特征基因集的基因表达量来分析、预测或评估受试者的癌症风险。
[0083]
本领域技术人员能够理解,可以联合使用任何能够反映tat细胞的富集程度的指标和能够反映tat特征的基因集的基因表达的富集情况的指标来分析、预测或评估受试者的癌症风险。
[0084]
本文引用的所有公开文件、专利和专利申请通过引用的方式全文并入本文。
[0085]
实施例
[0086]
本发明已经通过下述实施例进行了说明,但应当理解的是,上述实施例只是用于举例和说明的目的,而非意在将本发明限制于所描述的实施例范围内。对于本领域技术人员来说,本发明可以有各种更改和变化,包括技术特征的组合、重组。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
[0087]
实施例1:确定tat细胞及tat-tcr序列
[0088]
发明人从immuneaccess公开数据库收集到了百余例肿瘤组织/外周血配对的tcr测序数据并依据cdr3序列信息对所有的tcr克隆划分成了不同的亚群。随后发明人从immuneaccess数据库收集到了500余例正常健康人的pbmc样本的tcr测序数据。从pbmc(外周血单个核细胞)和til(肿瘤浸润的t细胞)中均出现的tcr序列中排除上述正常健康人pbmc样本中出现的tcr序列,将由此得到的tcr序列界定为tat-tcr序列。将包含该tat-tcr序列的t细胞界定为tat细胞(图1)。
[0089]
实施例2:使用tat-tcr序列评估癌症风险
[0090]
发明人按照如下图所示的数据过滤和筛选步骤(图1),构建了基于tat-tcr的二分类预测模型,用于预测每一条tcr是肿瘤相关tcr的概率。
[0091]
随后发明人设计了一个tcr repertoire risk score(trrs)用于定量评估病人外周血中的肿瘤相关t淋巴细胞的富集程度,具体计算方式为:对于每一个病人样本,发明人利用其构建好的二分类模型对其pbmc中测到的每一条tcr序列进行预测,得到该条tcr序列为tat的概率;随后在给定的概率阈值下(66%或75%)我们计算该病人中被预测为tat序列的数量a;接下来我们计算该病人样本pbmc中来源于正常人人群的tcr序列(通过上述公开数据库中获取到)的数量b,则trrs的计算方式为a/b。
[0092]
发明人发现trrs能够显著区分癌症病人和正常人,并且还能够区分癌症病人和病毒感染的人群(图2)。
[0093]
实施例3:分析tat细胞的克隆增殖能力
[0094]
发明人分析了不同t细胞亚群的克隆增殖情况,计算方式为利用lymphoseq r包(https://github.com/davidcoffey/lymphoseq)。
[0095]
发明人发现共享血液和肿瘤浸润的微环境中同时存在的tcr序列的t细胞亚群的克隆性增殖情况更高,尤其是pbmc来源的这类tat细胞亚群的克隆性增殖情况更高,表明这一类t细胞亚群(tat细胞)与肿瘤相关抗原的结合能力更强,具备更强的肿瘤杀伤活性。
[0096]
实施例4:确定tat特征基因集
[0097]
随后发明人借助于单细胞测序数据(scrna-tcr-seq)用于分析单细胞精度下tat细胞的基因转录组特征。结果表明,相对于非癌症相关t细胞(clonal或non-clonal t细胞),tat细胞中t细胞增殖、激活等基因模块的基因表达显著升高。
[0098]
通过公开数据整理,发明人收集到的14组肿瘤组织/外周血配对的scrna-tcr-seq数据,其pmid分别为:28622514、29942094、30479382、33651880、33514641、33531485、32103181、33504936、33861994、34179824、33900375、33651880、34290408。
[0099]
为了降低单细胞测序数据分析过程中的批次效应,发明人对每一组数据进行单独的差异表达分析,比较的两组分别为tat和nonclonal亚群(没有观察到克隆增殖情况的亚群)。
[0100]
发明人选取了在超过10组数据中(总计14组数据)都显著表达的基因,构成tat特征基因集。
[0101]
发明人发现在不同的数据集中检测到的tat特征基因能够独立预测其他数据集中的tat细胞(图4)。具体预测的计算方式为利用每一组数据做差异表达分析时得到的基因集,在该组数据集上训练一个区分tat和nonclonal细胞(属于非癌症相关t细胞)的二分类机器学习模型,并将该模型独立应用在其他13组数据集上去验证模型的表现。
[0102]
实施例5联合使用tat-tcr和转录组信息评估癌症风险
[0103]
随后,发明人结合tat-tcr预测模型和tat特征基因集,设计了一个肿瘤筛查的框架。获取到受试者血液样本后发明人分别测定该样本的tcr序列组库数据以及基因表达定量数据。
[0104]
tcr序列组库数据输入到发明人的tat预测模型中,获取trrs的打分;基于tat特征基因集计算基因表达定量数据中的转录组分数;随后这两方面的分数作为两个独立的特征输入到逻辑回归模型中,构建二分类模型;回归方程的相应变量的值作为肿瘤风险发生概率,用于评估该样本肿瘤发生的概率。
[0105]
发明人利用seurat r包(https://github.com/satijalab/seurat)中的addmodulescore去计算基于tat特征基因集的转录组特征分数。通过模型评估,发明人发现单独使用trrs分数或者基于tat特征基因集的转录组特征分数去区分肿瘤和正常人,模型的auc分别达到了0.675和0.673;当将两者结合起来构建一个二分裂的逻辑回归模型去区分肿瘤和正常人的时候,模型的auc达到了0.797,优于使用单一特征进行预测分析的结果。
[0106]
结果表明,可以单独使用tat-tcr序列、单独使用tat特征基因集的基因表达量或联合使用两者来预测受试者的癌症风险。结果表明,联合使用tat-tcr和tat特征基因集的基因表达量的预测结果的灵敏度和特异性要优于单独使用其中之一的预测结果。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。