一种检测crispr基因编辑脱靶的方法
技术领域
1.本发明涉及检测方法技术领域,特别是一种检测crispr基因编辑脱靶的方法。
背景技术:
2.crispr/cas9基因编辑系统是近年来研发成功的一种基因编辑系统,由于操作简单,快捷高效等优点,且其快速、准确的基因编辑能力,使通过基因定点突变治疗人类遗传疾病成为可能,已被广泛运用于基因工程。crispr/cas9系统包括了cas9核酸内切酶和grna,在crispr/cas9系统发挥功能的过程中,grna引导cas9蛋白识别靶点ngg序列,随后剪切dna双链,在编辑位点引进dsb(双链断裂),细胞通过招募自身修复蛋白,如h2ax、mre11等,修复dsb位点,从而实现对dna的定点编辑。然而受到技术限制,crispr/cas9系统在应用中存在脱靶效应,即切割与靶向序列相类似的非靶点区域的序列,脱靶效应的发生会破坏非目标基因的功能,这也限制了crispr/cas9系统在生物医学领域的运用。由于脱靶效应是crispr/cas9基因编辑过程中存在的潜在风险,因此需要对其进行检测,才能保证crispr/cas9基因编辑系统的正常应用。
3.现有技术中,crispr/cas9基因编辑系统脱靶检测的方法是主要全基因组测序法和guide-seq两种技术。其中,全基因组测序法是通过软件预测潜在的脱靶位点,然后将基因组测出来进行比对,该方法只能检测已修复后的位点,存在检测周期较长,灵敏度低的缺点。guide-seq技术主要是利用一种短双链寡核苷酸标签来标记crispr/cas9诱导的断裂(在靶和脱靶),然后对标签所在的基因区域进行高通量测序,该方法整个检测过程时间较长,步骤繁琐,且不能保证解决所有的断裂位点都被标记的问题。综上所述,提供一种能有效对crispr/cas9基因编辑系统脱靶效应进行检测的技术显得尤为必要。
技术实现要素:
4.为了克服现有crispr/cas9基因编辑系统脱靶检测的方法,由于技术限制,存在检测周期较长,灵敏度低的缺点,以及步骤繁琐,且不能保证所有断裂位点都被标记的弊端,本发明提供了通过对crispr/cas9基因编辑过程中cas9蛋白和靶位点和细胞自身修复蛋白h2ax、mre11与靶位点形成的蛋白-核酸复合体进行免疫共沉降,并结合测序及其他必要技术手段,实现对脱靶位点的筛选,极大地简化了识别脱靶效应的过程,同时也提高了结果的准确性,能为更好预测基因组编辑如何在临床环境中发挥的作用起到了有力技术支撑的一种检测crispr基因编辑脱靶的方法。
5.本发明解决其技术问题所采用的技术方案是:
6.一种检测crispr基因编辑脱靶的方法,其特征在于包括细胞交联、抗体固定、超声处理、染色质免疫共沉淀、dna纯化、dna处理、文库扩增七个步骤:所述细胞交联包括取样、孵育洗下、第一次离心去清、第二次离心去清、第三次离心去清、对细胞交联、终止交联、离心、第四次离心去清九个分步骤;所述抗体固定包括清洗磁珠、重悬磁珠及抗体两个分步骤;所述超声处理包括准备细胞样品及辅助反应液和抑制剂、超声、离心、分装、加蛋白酶k
水浴、加入dna清洗磁珠、酒精洗涤、测浓度并准备chip实验阳性对照样品八个分步骤;所述染色质免疫共沉淀包括收集抗体、洗涤、补充液体、免疫共沉淀共四个分步骤;所述dna纯化包括第一次去清、混匀水浴、涡旋、清洗、第二次去清、洗涤、取清液共七个分步骤;所述dna处理包括dna末端补平、adapter连接、纯化分选及洗脱三个分步骤;所述文库扩增包括pcr仪进行实验、分选纯化及洗脱dna两个分步骤。
7.进一步地,所述细胞交联,取样是以人细胞培养液系进行实验;孵育洗下中,先通过多个平板取部分人细胞培养液,每个平板加入胰酶然后孵育,用滴管吹吸将细胞充分洗下并进入下一工序;第一次离心去清中,加入剩余的人细胞培养液抑制胰酶消化,然后离心去清并进入下一工序;第二次及第三次离心去清中,分别先加入磷酸盐缓冲液重悬,然后去清并进入下一工序;对细胞交联中,通过甲醛溶液室温固定并旋转实现细胞的交联并进入下一工序;终止交联中,加入甘氨酸终止固定并旋转,实现终止交联并进入下一工序;第四次离心去清中,先将离心终止交联后产物用磷酸盐缓冲液洗两次,然后离心去清,所得产物用于超声处理步骤。
8.进一步地,所述抗体固定,清洗磁珠中,取免疫磁珠和磷酸盐缓冲液清洗2次,然后磁力架去清液并进入下一工序;重悬磁珠及抗体中,用磷酸盐缓冲液重悬清洗磁珠和抗体并混匀旋转,所得产物用于染色质免疫共沉淀步骤。
9.进一步地,所述超声处理,准备的细胞样品及辅助反应液、抑制剂包括细胞交联步骤中获得的细胞样品产物、十二烷基硫酸钠缓冲液、蛋白酶抑制剂,并将上述所有液体在冰上放置一段时间后进行超声并进入下一工序;超声后产物进行离心、然后取上清至新离心管中并进入下一工序;分装中,取部分超声后产物至新离心管,超声后两部分样品,一部分作为不进行免疫共沉淀实验,另外一部分作为免疫共沉淀、用于染色质免疫共沉淀步骤实验并进入下一工序;分装后产物加蛋白酶k水浴后,加入dna清洗磁珠后在室温放置一段时间并进入下一工序;酒精洗涤中,水浴后产物先去清液,然后用酒精洗涤两次并晾干进入下一工序;测浓度并准备chip实验阳性对照样品中,部分产物采用荧光定量仪测浓度,剩余留作实验阳性对照品。
10.进一步地,所述染色质免疫共沉淀,收集抗体中,取出抗体固定步骤预先结合的抗体-磁珠,通过磁力架收集抗体并进入下一工序;洗涤中,在磁力架上,用磷酸盐缓冲液将磁珠快速旋转洗涤两遍并进入下一工序;补充液体中,补充的液体是ripa 0.2m nacl、50xpi、超声处理步骤的部分产物dna清液、1xte,并将上述液体补充至1.4ml后进入下一工序;免疫共沉淀中,将洗涤分步骤产物抗体-beads通过磁力架去清液后,然后加入补充液体分步骤的清液,混匀旋转过夜。
11.进一步地,所述dna纯化,第一次去清中,将染色质免疫共沉淀步骤获得的产物放置于磁力架去清液并进入下一工序;混匀水浴中,具体通过缓冲液、十二烷基硫酸钠缓冲液、蛋白酶k重悬第一次去清所得产物,然后混匀水浴并进入下一工序;涡旋后将磁力架取清液至新的试管内并进入下一工序;清洗中,采用磁力架清洗产物,并合并涡旋、第一次去清分步骤的清液进入下一工序;第二次去清中,对清洗工序后产物加1
×
clean beads通过磁力架去清液并进入下一工序;洗涤中,通过两次洗涤产物、晾干后进入下一工序;取清液中,通过缓冲液溶解洗涤后产物的dna,混匀放置一段时间后短暂离心在磁力架上取清液。
12.进一步地,所述dna处理,dna末端补平中,将超声处理或dna纯化后产物、末端补平
缓冲液、无核酸酶水使用移液器吹打混匀,并离心将反应液收集至试管底,在pcr仪中进行实验;adapter连接中,将细胞交联后产物、引物接头连接缓冲液、dna连接酶、引物接头使用移液器混匀,并离心将反应液收集至试管底,在pcr仪中进行实验;纯化分选及洗脱中,用dna clean beads对adapter连接所得反应产物进行纯化,然后单轮分选,用水洗脱dna。
13.进一步地,所述文库扩增,pcr仪进行实验中使用的原料是pcr引物混合物3、保真扩增混合物、dna处理步骤所得的连接产物;分选纯化及洗脱中,用0.6
×
、0.35
×
dna clean beads对细胞交联步骤产物进行双轮分选纯化,然后用水洗脱dna,所得产物最终用于高通量测序。
14.本发明有益效果是:本发明在多个步骤共同作用下,通过对crispr/cas9基因编辑过程中cas9蛋白和靶位点和细胞自身修复蛋白h2ax、mre11与靶位点形成的蛋白-核酸复合体进行免疫共沉降,并结合测序及其他必要技术手段,实现对脱靶位点的筛选,极大地简化了识别脱靶效应的过程,同时也提高了结果的准确性,能为更好预测基因组编辑如何在临床环境中发挥作用起到了有力技术支撑。克服了现有crispr/cas9基因编辑系统脱靶检测的方法,由于技术限制,存在检测周期较长,灵敏度低的缺点,以及检测过程时间较长,步骤繁琐,且不能保证所有断裂位点都被标记的缺点。基于上述,本发明具有好的应用前景。
附图说明
15.以下结合附图和实施例将本发明做进一步说明。
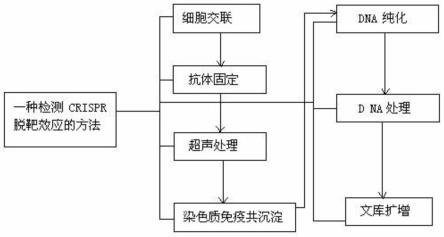
16.图1是本发明流程示意图。
具体实施方式
17.图1所示,一种检测crispr基因编辑脱靶的方法,包括细胞交联、抗体固定、超声处理、染色质免疫共沉淀、dna纯化、dna处理、文库扩增七个步骤,具体步骤如下。
18.图1所示,细胞交联包括如下分步骤,(1):以人细胞系u20s进行实验,人细胞系是现有技术利用crispr/cas9系统获得的基因编辑细胞系。(2):取血清培养液6-8ml(毫升),其余弃去(血清培养液是分步骤(1)的细胞培养液),然后将血清培养液滴在每个(三-四个)实验器材平板上并加入2ml左右胰酶,37℃温度条件下孵育1min,用滴管吹吸将细胞充分并洗下,使细胞消化成单细胞形态。(3):将步骤(2)获得的细胞产物经试管加入之前的血清培养液(新的血清培养液),抑制胰酶的消化(也就是结束消化),通过离心机在300g离心力条件下离心10min,并去试管内清液(去清)。(4):将步骤(3)产物用5ml 1
×
pbs(磷酸盐缓冲液)重悬,通过离心机在300g离心力条件下离心10min,去清。(5):将步骤(4)产物用10ml 1
×
pbs重悬,在300g离心力条件下离心10min,去清。(6):对步骤(5)产物用1%甲醛溶液室温固定10min,然后旋转试管(该步骤实现细胞交联,保持细胞蛋白与dna的相互作用的状态)。(7):将步骤(6)产物加入1ml 2m(摩尔每升)甘氨酸终止固定5-10min,然后旋转试管(该步骤实现终止交联)。(8):对步骤(7)中产物在300g离心力条件下离心5min。(9):对步骤(8)产物用5ml 1
×
pbs洗两次,离心去清(获得的产物进入超声处理步骤)。
19.图1所示,抗体固定包括如下分步骤,(1):将30ul(微升)dynabeads(免疫磁珠)用600ul 1
×
pbs(磷酸盐缓冲液)清洗2次,然后置于磁力架去清液(该步骤目的是清洗磁珠)。(2):用150ul 1x pbs重悬磁珠和抗体(抗体是cas9、h2ax、mre11,抗体量具体看说明书,抗
体购自生产和销售抗体的生物公司)在转速2-6hr、温度4℃条件下混匀旋转(该步骤为抗体准备过程,抗体用于染色质免疫共沉淀步骤实验)。
20.图1所示,超声处理包括如下分步骤,(1)将细胞样品(步骤细胞交联所得产物) 700ul(可调整用量)0.5%sds(十二烷基硫酸钠缓冲液) pi(蛋白酶抑制剂)置于试管内在冰上放置15min。(2):将步骤(1)产物用超声波粉碎设备超声25cycles(该步骤目的是打断dna片段至200-500bp)。(3):将步骤(2)产物用离心机离心力12000g、温度4条件下离心10min,并取上清至新离心管中。(4):将步骤(3)产物取50ul至新离心管作为input(超声后的样品分为两部分,一部分作为input(不进行免疫共沉淀),另外一部分作为ip(免疫共沉淀)用于染色质免疫共沉淀步骤进行免疫共沉降实验)。(5);将步骤(4)另外一部分所得产物加入5ul protein k(蛋白酶k),在55℃温度条件下水浴2hr(小时)(该步骤作用是消化蛋白)。(6):将步骤(5)获得的产物加入提前放至室温的1x dna clean beads(抗体固定步骤获得的dna清洗磁珠)室温放置5min。(7):将步骤(6)获得的产物用mpc5min去清液,然后用500-600ul80%现配酒精洗涤两次,并置于磁力架,5-15min晾干进入下一步骤。(8):将步骤(7)获得的产物用50ul0.1
×
te(缓冲液)混匀,放置5min,采用离心机短暂(5秒钟)离心后、置于磁力架上取清液1ul用作qubit(荧光定量仪)测浓度,剩余留作input(input作为chip实验的阳性对照)。
21.图1所示,chromatin immunoprecipitation(染色质免疫共沉淀)包括如下分步骤,(1)取出抗体固定步骤所获得预先结合的抗体-beads(磁珠),通过磁力架放置抗体。(2):在磁力架上,用1mlpbs将步骤(1)磁珠快速旋转洗涤两遍。(3):将(ripa 0.2m nacl)688ul 14ul(50xpi) dna清液(抗体固定步骤所获得的产物) 1xte,并将试管内液体补至1.4ml。(4):将步骤(2)中抗体-beads在磁力架上去清液后,然后加步骤(3)清液,在4℃温度条件下混匀旋转过夜(该步骤得到ip(免疫共沉淀)产物,也就是获得目的dna片段,应用于dna纯化步骤)。
22.图1所示,dna纯化包括如下分步骤,(1):将染色质免疫共沉淀步骤所获的产物置于磁力架去清液,具体操作分别用1ml a/b/c/d buffer(缓冲液)&1ml 1x te清洗5轮,每轮清洗中,温度4℃条件下用离心机旋转5-10min,短暂(5秒钟左右)离心在磁力架上去清。(2)将步骤(1)的产物加入100ul 1
×
te(缓冲液)、3ul 10%sds(十二烷基硫酸钠缓冲液)、5ul proteinase k(蛋白酶k)重悬beads,在温度55℃条件下水浴≥2hr混匀。(3):将步骤(2)产物短暂(5秒钟左右)涡旋,然后在磁力架上取清液至新的ep管内。(4):将步骤(3)产物用100ul(90ul 1
×
te 10ul 5m nacl)在磁力架上清洗beads,并合并涡旋、第一次去清分步骤的清液进入下一工序。(5):将步骤(4)产物加1
×
clean beads室温放置5min,然后至于磁力架2min后去清液。(6):把步骤(5)产物用80%现配酒精洗涤两次(置于磁力架),放置5-15min后晾干。(7):将步骤(6)产物用30ul 0.1
×
te溶解dna,混匀放置5min后,在短暂(5秒钟)至于离心磁力架上取清液。(获得纯化过的dna,用于dna末端补平)
23.图1所示,dna处理包括如下分步骤,(1):dna末端补平,将dna(超声处理步骤获得的iuput或dna纯化步骤获得产物)、end prep mix 4(末端补平缓冲液)7.5ul、nuclease-free water(无核酸酶水to 32.5ul,使用移液器轻轻吹打混匀(勿振荡混匀),并短暂离心(5秒钟)将反应液收集至试管底,在20℃、15min,65℃、15min,4℃、hold(保持在4℃)条件分别在pcr仪中进行实验(dna末端补平)。(2)adapter连接,将dna末端补平后产物32.5ul、
rapid ligation buffer 2(引物接头连接缓冲液)12.5ul、rapid dna ligase(dna连接酶)2.5ul、dna adapter x(引物接头)2.5ul,使用移液器轻轻吹打混匀(勿振荡混匀),并短暂离心将反应液收集至管底,在在20℃、15min,4℃、hold条件分别在pcr仪中进行实验(链接引物接头)。(3):用1x dna clean beads对步骤(2)所获反应产物进行纯化,单轮分选,并用22ul水洗脱dna(dna处理步骤主要为dna末端修复和接入测序引物接头,用于高通量测序测序)。
24.图1所示,文库扩增包括如下分步骤,(1)将pcr primer mix 3for illumina(pcr引物混合物3,购自生物公司)5ul、vahts hifi amplification mix(保真扩增混合物)25ul、连接产物20ul(dna处理步骤所获得的产物),分别在95℃、3min,98℃、20s,60℃、15s,72℃、30s,72℃、5min,4℃、hold(保持在4℃)条件下,分别在pcr仪中进行实验(其中60℃、15s条件下进行12-15cycle(循环)实验)。(通过pcr扩增获得用于测序的文库)(2):先用0.6
×
dna clean beads对pcr扩增产物进行纯化、取上清至新的离心管、再用0.35
×
dna clean beads进行纯化、50ul洗脱液(10mm tris,ph=8-8.5)洗脱dna用于高通量测序(通过测序分析是否发生脱靶)。
25.图1所示,本发明在细胞交联、抗体固定、超声处理、染色质免疫共沉淀、dna纯化、dna处理、文库扩增步骤共同作用下,通过对crispr/cas9基因编辑过程中cas9蛋白和靶位点和细胞自身修复蛋白h2ax、mre11与靶位点形成的蛋白-核酸复合体进行免疫共沉降,并结合测序及其他必要技术手段,实现对脱靶位点的筛选,极大地简化了识别脱靶效应的过程,同时也提高了结果的准确性,能为更好预测基因组编辑如何在临床环境中发挥作用起到有力技术支撑。现有的guide-seq技术应用了一段特定序列识别脱靶位点,实际操作中需要将特定序列转染进入细胞系,因此检测拖把的效率比较低,本发明利用染色质免疫共沉淀获得脱靶位点dna片段,相较于现有技术过程简单,准确性高,克服了现有crispr/cas9基因编辑系统脱靶检测的方法,由于技术限制,存在检测周期较长,灵敏度低的缺点,以及检测过程时间较长,步骤繁琐,且不能保证所有断裂位点都被标记的缺点。
26.以上显示和描述了本发明的基本原理和主要特征及本发明的优点,对于本领域技术人员而言,显然本发明限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。
27.此外,应当理解,虽然本说明书按照实施方式加入以描述,但并非实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。