1.本发明涉及植物基因工程技术领域,具体涉及一种受黑暗和绿光诱导的植物诱导型启动子及其应用。
背景技术:
2.植物基因启动子是rna聚合酶识别、结合和开始转录的一段dna序列,它含有rna聚合酶特异性结合和转录起始所需的保守序列。植物的生长发育和生长周期是不同基因在时间和空间上有序表达的结果,这些基因表达调控的关键环节之一是在特异启动子调控下的转录水平的调控。植物组织特异型或条件诱导型启动子是合理分配能量流向和环境适应性的主要因素。
3.目前基因工程技术中采用的启动子包括组成型启动子和诱导型启动子。组成型启动子能够驱动目的基因在植物各组织中持续表达,但是会过度消耗受体细胞内的物质和能量,打破植物自身的自然规律,往往不能很好的调控基因的表达,因此在应用中存在一定的缺陷。在已发表生物自发光植物的设计中,研究者利用组成型启动子35s驱动真菌发光系统基因的表达,创造出发出波长约为520nm的烟草,khakhar a等将构巢曲霉(aspergillus nidulans)基因组中的npga(4
’‑
phosphopantetheinyl transferase)基因、光茸菌(neonothopanus nambi)基因组中的h3h(hispidin-3-hydroxylase)基因和hisps(hispidin synthase)基因,以及真菌荧光素酶(luz)基因整合到moclo质粒中,这些基因均使用组成型启动子35s驱动这些基因的表达(mitiouchkina et al.,2020)。由于引入的发光系统是组成型启动子,该烟草会持续发出绿光,白天也会消耗植物体内大量能量,可能会对植物的生长发育或抗逆性产生不良影响。
4.诱导型启动子在受外源物理、化学因素等的诱导下,能快速诱导基因转录的“开”与“关”,因此可根据实验需要调控转基因在植物中的表达。目前应用较为广泛的诱导表达系统有:四环素类诱导表达系统、类固醇类诱导系统、地塞米松诱导系统、激素类诱导表达系统以及杀虫剂和乙醇表达系统。这些化学诱导启动子的反应条件需要人为添加化学诱导物质才能激活目的基因的表达,控制较难且效率低,在一定程度上可能对植物产生毒害。因此,开发天然诱导型启动子已成为当前基因工程的必要需求(kusunoki&yamamoto,2017)。
5.在高等植物的生长发育过程中,光除了参与光合作用积累生物量,还作为一种重要的信号调控相关基因的表达。大量研究表明,植物体内存在一系列受光诱导表达的内源基因。因此,对这些基因的启动子进行研究,开发光诱导型启动子诱导基因表达系统对植物基因工程研究和应用具有重要意义。目前对黑暗诱导型启动子没有进行系统的挖掘和利用,开发具有应用价值的黑暗诱导启动子,诱导发光植物在夜间发光,对自发光植物的开发和选育具有重要意义。
技术实现要素:
6.本发明的目的在于提供了一种受黑暗诱导的启动子元件,在黑暗条件下能够诱导
下游靶基因的表达,而在自然光下不能诱导下游靶基因表达。利用该元件控制植物发光系统模块只在黑暗时发光,使得发光植物白天积累能量晚上释放光芒,进一步提高植物的发光强度。
7.为实现上述目的,本发明采用如下技术方案:
8.本发明通过对油菜黑暗处理转录组数据进行分析,筛选出一个黑暗诱导启动子,其核苷酸序列如seq id no.1所示。研究表明,相较于正常光照条件,该启动子在黑暗或绿光条件下诱导下游基因的表达量提高10倍以上。
9.本发明提供了seq id no.1所示的dna分子的获取方法,包括:首先提取油菜基因组dna,然后利用pcr技术克隆所述的dna分子片段;pcr扩增采用的引物为:f引物:5
’‑
ttattgcctaagattgatg-3’,r引物:5
’‑
ttttcttatttaatgcaaagt-3’。
10.本发明对上述启动子序列做核心序列分析,推测547-551位以及726-731位的cacgtg为黑暗诱导的核心区域。进一步的,本发明对上述启动子序列进行改造,在seq id no.1所示的核苷酸序列中在光诱导核心元件g-box(cacgtg)附近200bp范围内重复插入若干个核心元件。
11.具体的,在核苷酸序列如seq id no.1所示的dna分子序列的第552-725位之间插入若干个包含核心元件片段,所述核心元件的序列为5
’‑
cacgtg-3’。
12.作为优选,插入片段的选取除了核心元件,还包含核心元件上下游的5~6个碱基。具体的,所述包含核心元件片段的序列为5
’‑
gataccacgtgtatgac-3’和/或5
’‑
atacaacacgtggcaaca-3’。
13.上述两种片段在seq id no.1所示的核苷酸序列的第558-719位点之间插入,每种片段重复1-2次。
14.作为优选,改造后的启动子的核苷酸序列如seq id no.2所示。研究表明,改造后的启动子在黑暗或绿光条件下对下游基因的诱导型显著增强至40倍以上。
15.本发明提供了包含所述启动子的相关的生物材料,为下述a1)-a3)中的任意一种:
16.a1)含有所述的启动子的基因表达盒;
17.a2)含有所述的启动子的重组载体。
18.a3)含有所述的启动子的重组细胞。
19.本发明提供了所述的启动子在作为植物诱导型启动子中的应用,所述诱导型启动子为黑暗诱导型和/或绿光诱导型启动子。
20.进一步的,所述植物为烟草、蝴蝶兰、油菜、水稻、菊花植物等草本植物。
21.本发明还提供了所述的启动子在植物体内启动目的基因表达的应用。通过在目的基因上游整合所述的启动子,可以使得目的基因在黑暗或绿光条件下被诱导表达。
22.本发明提供了一种自发光植物的培育方法,包括:将所述的启动子和自发光基因元件导入受体植物中,得到自发光基因受黑暗诱导和/或绿光诱导表达的植物。
23.进一步的,所述自发光植物的培育方法包括以下步骤:
24.(1)利用多基因组装技术将hisps基因、cph基因、h3h基因、npga基因和luz基因整合到受体载体中,构建获得多基因载体,所述多基因载体中luz基因的上游整合有所述的启动子;
25.(2)利用转基因技术将多基因载体中的目标基因片段导入受体植株中,培育,获得
生物自发光受黑暗诱导和/或绿光诱导的转基因植株。
26.其中hisps的序列信息见基因登录号:qjq48095.1;cph的序列信息见基因登录号:qjq48093.1;h3h的序列信息见基因登录号:qjq48094.1;npga的序列信息见基因登录号:qjq48097.1;luz的序列信息见基因登录号:qjq48096.1;bnc3h的序列信息见基因登录号:xp_009133464.2。
27.进一步的,步骤(1)中,采用transgene stacking ii系统进行多基因组装。以pyl322d1作为供体载体ⅰ,pyl322d2作为供体载体ⅱ,pyltac380gw作为受体载体。
28.具体的,所述多基因载体的构建方法包括以下步骤:
29.1)将hisps基因片段、h3h基因片段分别插入供体载体pyl322d1的多克隆位点中获得供体载体pyl322d1-35s-hisps,pyl322d1-35s-h3h,将带有所述的启动子的luz基因片段替换供体载体pyl322d1的35s启动子获得供体载体pyl322d1-edie-luz;
30.将cph基因片段和npga基因片段分别插入供体载体pyl322d2的多克隆位点中获得供体载体pyl322d2-35s-cph,pyl322d2-35s-npga;
31.2)将供体载体pyl322d1-35s-hisps和受体载体pyltac380gw按1:1至2:1混合,共转入大肠杆菌ns3529感受态细胞中,涂布于含卡那霉素和氯霉素的双抗培养基中培养,取阳性菌株提取质粒;
32.3)使用归巢酶i-sce i对步骤2)提取的质粒进行酶切,再转化大肠杆菌菌株neb10-β,培养,筛选,提取质粒,获得含有目的基因hisps的阳性克隆pyltac380gw-hisps;
33.4)将供体载体pyl322d2-35s-cph和步骤3)制备的受体载体pyltac380gw-hisps按1:1至2:1混合,共转入大肠杆菌ns3529感受态细胞中,涂布于含卡那霉素和氨苄霉素的双抗培养基中培养,取阳性菌株提取质粒;
34.5)使用归巢酶pi-sce i对步骤4)提取的质粒进行酶切,再转化大肠杆菌菌株neb10-β,培养,筛选,提取质粒,获得含目的基因hisps和cph的阳性克隆pyltac380gw-hisps-cph;
35.6)重复步骤2)-5),以上一步骤获得的含有目的基因的新质粒作为受体载体,交叉使用含不同基因的d1、d2供体载体进行重组,直至所有目的基因组装到受体载体上,最后一步bp重组反应连入去除筛选标记基因表达盒元件,构建得到多基因载体pyltac380gw-hisps-cph-h3h-npga-edie-luz。
36.步骤(2)中,将构建的多基因片段导入受体植株中,使其在植株体内表达,表达的各蛋白酶参与咖啡酸循环,其中luz基因只在黑暗或绿光条件下诱导表达,使得植物只在黑暗时发光。
37.进一步的,采用农杆菌介导技术将多基因片段导入受体植株。所述农杆菌采用eha105。
38.进一步的,所述受体植株为烟草、蝴蝶兰、油菜、水稻或菊花植物。
39.进一步的,所述hisps基因的编码序列如seq id no.3所示,所述cph基因的编码序列如seq id no.4所示,所述h3h基因的编码序列如seq id no.5所示,所述npga基因的编码序列如seq id no.6所示,所述luz基因的编码序列如seq id no.7所示。
40.本发明具备的有益效果:
41.(1)本发明首次从油菜中鉴定获得如seq id no.1所示的启动子序列,该启动子为
黑暗诱导型和/或绿光诱导型启动子。该启动子对植物基因工程研究和应用具有重要作用,其应用前景十分广阔。
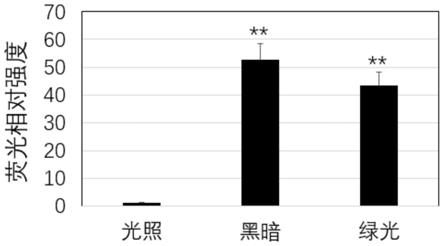
42.(2)本发明改造获得的启动子在黑暗和绿光照射下对下游报告基因的诱导性显著增强。
43.(3)本发明利用所述启动子构建生物自发光系统模块,成功诱导植物在夜间发光,为发光植物白天积累能量晚上释放光芒,进一步提高植物的发光强度提供有用的启动元件。
附图说明
44.图1为黑暗诱导启动子die瞬时转化载体示意图。
45.图2为黑暗诱导启动子die诱导报告基因luz强度测定。
46.图3为黑暗诱导启动子edie瞬时转化载体示意图。
47.图4为黑暗诱导启动子edie诱导报告基因luz强度测定。
48.图5为黑暗和绿光诱导型转基因载体380gw-edie-g6构建过程中各轮酶切检测。
49.图6为qpcr检测黑暗和绿光诱luz基因的表达。
50.图7为黑暗和绿光诱导型转基因烟草苗期检测。
51.图8为黑暗和绿光诱导型转基因烟草苗期荧光强度分析。
具体实施方式
52.下面结合具体实施例对本发明做进一步说明。以下实施例仅用于说明本发明,不用来限制本发明的适用范围。在不背离本发明精神和本质的情况下,对本发明方法、步骤或条件所做的修改或替换,均属于本发明的范围。
53.下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
54.实施例1黑暗诱导型启动子的鉴定
55.1、通过对油菜黑暗处理转录组数据进行分析,筛选出一个黑暗诱导启动子(dark induce element,die),其核苷酸序列如seq id no.1所示。
56.利用pcr技术扩增出该启动子序列,所用f引物:5
’‑
ttattgcctaagattgatg-3’,r引物:5
’‑
ttttcttatttaatgcaaagt-3’,将扩增出的启动子元件连入启动子分析载体(图1),测序验证正确无误后,转入农杆菌eha105中,将含有已验证正确的载体质粒的eha105菌液分别在la kana rif板上划线活化,28℃培养36h,从板上挑取菌落,转入lb kana rif 15μm as培养基中,28℃,200rpm培养至od
600
=0.8-1.0,4000rpm,10min收集菌体,用侵染液(含10mm mgcl,10mm mes,150μm as)悬浮农杆菌菌体,室温静置2~3h。
57.2、烟草叶片中瞬时表达验证,用1ml的针头在烟草叶片表面轻轻点开一个小口,再用去掉针头的针管吸取菌液,从烟草叶片伤口处注射进入叶片。在正常白光照培养箱中(25℃,12h光照\12h黑暗,6000lux)培养48h后,光照处理为12h光照后继续光照3h,黑暗处理是光照12小时后开始3h黑暗,绿光处理是12h光照后改为绿色荧光灯照射3h,之后取菌液侵染叶片样,液氮磨样,取0.02g加入100μl裂解缓冲液(promega,luciferase assay system提供),混匀30s,冰上放置。加入5μl细胞裂解物到酶标板中,加入45μl底物(promega,
luciferase assay system),混匀2s,10s后用酶标仪检测测量发出荧光的信号值。
58.结果如图2所示,黑暗或绿光3h处理可以显著诱导报告基因的表达至10倍以上,确定启动子为黑暗诱导型。
59.实施例2黑暗诱导型启动子的改造
60.我们对实施例1筛选到的dark induce element(die)启动子做核心序列分析,根据已有结果表明该启动子部分有核心元件gbox(cacgtg),可能是黑暗诱导的核心区域,我们对黑暗诱导核心元件gataccacgtgtatgac和atacaacacgtggcaaca做多重重复设计,该启动子元件命名为engineered dark induce element(edie),其核苷酸序列如seq id no.2所示。
61.根据上述序列委托公司合成相应的启动子片段,连入启动子分析载体(图3),测序验证正确无误后,转入农杆菌中,通过烟草叶片瞬时转化实验(操作方法同实施例1)验证。
62.结果如图4所示,改造后的edie启动子在黑暗和绿光照射下对下游报告基因的诱导性显著增强至40倍以上,表明改造后的edie元件,可以应用于需要黑暗或者绿光诱导的目的基因的表达。
63.实施例3黑暗诱导型发光植物转基因载体构建
64.根据上述结果,使用改造后的黑暗增强型启动子元件edie驱动植物自发光系统中luz基因的表达,该操作中使用华南农业大学刘耀光院士课题组开发的transgene stacking ii系统进行多基因组装,参见申请号为2017103841977的中国专利,pyl322d1、pyl322d2和pyltac380gw为华南农业大学刘耀光教授实验室馈赠。
65.将构巢曲霉(aspergillus nidulans)基因组中的npga(4
’‑
phosphopantetheinyl transferase)基因、光茸菌(neonothopanus nambi)基因组中的h3h(hispidin-3-hydroxylase)基因和牛奶树碱合成酶基因hisps(hispidin synthase),咖啡酰丙酮酸水解酶基因cph(caffeoyl pyruvate hydrolase),真菌荧光素酶(luz)基因整合到pyltac380gw质粒中,其中npga、h3h、hisps、cph均由35s启动子驱动,luz由黑暗诱导型启动子edie启动子驱动,最后通过一步bp重组反应连入去除筛选标记基因表达盒元件,从而构成黑暗诱导型载体pyltac380gw-hisps-cph-h3h-npga-edieluz(pyltac380gwedie-g6)。
66.所述hisps基因的编码序列如seq id no.3所示,所述cph基因的编码序列如seq id no.4所示,所述h3h基因的编码序列如seq id no.5所示,所述npga基因的编码序列如seq id no.6所示,所述luz基因的编码序列如seq id no.7所示。
67.具体过程如下。
68.(1)构建供体载体,pyl322d1-35s-hisps,pyl322d2-35s-cph,pyl322d1-35s-h3h,pyl322d2-35s-npga,pyl322d1-edie-luz。
69.各目标基因片段委托生物公司合成,hisps、h3h基因片段分别插入pyl322d1的多克隆位点获得pyl322d1-35s-hisps,pyl322d1-35s-h3h;cph、npga基因片段分别插入pyl322d2的多克隆位点获得pyl322d2-35s-cph,pyl322d2-35s-npga;edie启动子与luz基因片段一起合成,利用同源重组技术替换pyl322d1基础载体中的35s启动子,从而构建成pyl322d1-edie-luz。
70.(2)将供体载体pyl322d1-35s-hisps和受体载体pyltac380gw(按1:1至2:1)混合在ns3529感受态中进行共转,采用热激法,冰浴30min,热激90s,冰浴2-3min,在不含抗生素
的lb中,37℃,200rpm,2h复活,涂在含kanamycin(km,25mg/l)和chloramphenicol(chl,15mg/l)的la板上,约18h后长出单克隆,用ddh2o将所有单克隆冲洗至管中,抽提混合质粒。
71.(3)取100-200ng混合质粒用0.5ul i-sce i(neb)在10ul体系中酶切4-5h,转化大肠杆菌菌株neb10-β(博迈德),涂在含kanamycin(km,25mg/l)的la板上,37℃,15h后挑单克隆,在lb(含25mg/l km和0.5mm iptg)中培养,并进行菌液pcr鉴定,使用green taq mix,将能扩增出亮带的进一步抽提质粒,各取200ng使用0.2ul not l在20ul反应体系中酶切验证,出现五条带,含目的基因6.2k bp以及1290、1447bp、1532bp、13816bp大小的四条骨架条带,即为所需阳性克隆pyltac380gw-hisps(图5,380gw-g1)。
72.(4)将供体载体pyl322d2-35s-cph和(3)中受体载体pyltac380gw-hisps(按1:1至2:1)混合在ns3529感受态中进行共转,按照(2)方法转化,涂在含kanamycin(km,25mg/l)和ampicillin(amp,70mg/l)的la板上,约18h后长出单克隆,用ddh2o将所有单克隆冲洗至管中,抽提混合质粒。
73.(5)取100-200ng混合质粒用0.5ul pi-sce i(neb),加0.5ul bsa在10ul体系中酶切4-5h,随后按照(3)中方法进行转化及验证,出现六条带,含目的基因1.7k bp cph与6.2k bp hisps大小条带即为阳性克隆pyltac380gw-hisps-cph(图5,380gw-g2)。
74.(6)更多回合的重组,交叉使用含不同基因的d1、d2供体载体与上一轮构建完成的受体载体进行共转,构建完成pyltac380gw-edie-g6,最后通过bp反应,25℃,将pyltac380gw-edie-g6(200ng)与pylmfh-bnmlpro(100ng)在5μl反应中用1μl的5
×
bp酶混合物混合5小时。然后加入1μl proteinase k溶液以在37℃下终止反应10分钟。转入neb10-β(博迈德)感受态,挑单克隆鉴定即可。从单克隆菌株中抽提质粒酶切检测如图5,确认正确菌株用于后续转基因实验。
75.实施例4黑暗诱导型转基因发光植物表型鉴定
76.1、将含有已验证正确的载体质粒pyltac380gw-edie-g6的eha105菌液在la rif kana平板上划线,28℃、36h,挑单克隆至3-5ml lb培养基中200rpm,28℃,36h,按照1:100-1:50的比例扩大培养50ml 3-5h至od=0.6,然后将菌液离心,用ms0液体培养基(ms 3%sucrose ph5.8,50ml)悬浮菌体至od=0.6用于侵染。
77.2、选取野生型烟草zy100种子,在无菌ms培养基上种植4-5周至完全展开的烟草健康叶片,用手术刀切成0.5cm见方大小(切掉叶缘避开主脉),叶片上表面朝下在ms1固体培养基(ms 0.5mg/l iaa 2.0mg/l ba 3%sucrose 0.6-0.8%phytagel,ph=5.8)上,25℃暗培养2-3天。
78.3、将预培养过的烟草叶片加入到菌液中,涡旋振荡确保叶片切口被菌液浸没,静止5-30min,用无菌滤纸吸去附着的菌液;将侵染过的叶片上表面朝下置于ms1固体培养基上28℃、暗培养2d;将叶片上表面朝上放入含有timentin和草甘膦的ms1筛选培养基上,25℃光培养;当叶缘长出芽并可以分离时(1cm以上),将芽切下转移至含有抗生素(tm basta)的ms2(ms 0.5mg/l iaa 3%sucrose 0.6-0.8%phytagel,ph=5.8)固体培养基中,两周后长出根,打开育苗盒的盖子练苗一周后转入种植土中培养为t0代,t0代转基因植株收种。
79.用t0代转基因植株种子为t1代,取t1代种子发芽,在正常白光照培养箱中(25℃,12h光照\12h黑暗,6000lux)生长15天后,光照处理为12h光照后继续光照3h,黑暗处理是光照12小时后开始3h黑暗,绿光处理是12h光照后改为绿色荧光灯照射3h,之后取叶片样,液
氮磨样抽提总rna做反转录,和qpcr分析,确定黑暗诱导的luz表达受到黑暗和绿光诱导表达(图6)。部分t1代植株在全自动发光检测系统下拍照分析,发现转基因发光烟草在黑暗或者绿光诱导后发光亮度显著增强(图7),通过发光强度分析发现生物自发光烟草在黑暗或者绿光诱导后发光亮度显著增强至40倍以上(图8)。
80.以上结果表明该edie启动子在黑暗诱导和绿光诱导应用中较好的应用前景。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。