1.本发明属于有机合成药物化学领域,具体涉及一种瑞德西韦有关物质i及其制备方法。
背景技术:
2.在药品生产过程中,杂质研究是不可或缺且非常重要的一部分,药物中间体杂质残留会对后期药物成品带来潜在性风险。这些杂质的存在不仅影响药物的药效,而且还会产生生产储藏过程中的问题,部分杂质甚至会产生毒副作用。所以,对药物杂质进行分析和研究既可以保证用药的安全、有效、稳定,同时也为生产、流通过程的质量保证提供依据。杂质标准品是指用于杂质的鉴别、检查、含量测定的标准物质。所以,在生产和质量控制过程中,制备和研究杂质标准品是非常有必要的工作。
[0003][0004]
瑞德西韦属于核苷类似物,是rna依赖的rna聚合酶(rdrp)抑制剂,可以通过抑制病毒核酸合成抗病毒。目前针对埃博拉病毒感染的临床研究进行到了ii期阶段。感染mers的小鼠接受这种联合疗法后表现要好得多,病毒复制减少,肺功能改善。近期研究表明,瑞德西韦对抑制新冠病毒有一定活性作用。
[0005][0006]
吉利德公司在专利cn105343098a中公开的瑞德西韦合成方法是以碘代氨基三嗪与核苷内酯为起始物料,碘代三嗪和核苷对接形成关键中间体p3,进一步氰基取代等一系列转化生成瑞德西韦。
[0007]
发明人在瑞德西韦原料药的检测中,发现存在一种未知的杂质,其峰面积比例一般均在0.05-0.10%。经过高分辨质谱及核磁共振表征,该杂质为瑞德西韦有关物质i,即(2r,3r,4r)-1,1-二(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-2,3,5-三苄氧基戊烷-1,
4-二醇,其结构如下:
[0008]
在瑞德西韦分析方法开发及后续的成品检验过程中,需要用该杂质的标准品对瑞德西韦中此杂质进行定位及定量研究,因此需要用到大量的瑞德西韦杂质i的标准品。为了制备瑞德西韦有关物质i,可以采用传统的杂质分离方法:在瑞德西韦原料药样品中分离目标杂质,但是目标杂质经hplc(高效液相色谱法)检测,其含量最高只能达到0.11%,并且样品还有很多个相似含量的类似杂质,会对分离造成干扰,分离周期长,需要多次分离才能得到纯品,总收率一般只有0.03%左右。
[0009]
基于此,本技术提供了一种瑞德西韦有关物质i及其制备方法。
技术实现要素:
[0010]
本发明的目的是提供一种瑞德西韦有关物质i,即(2r,3r,4r)-1,1-二(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-2,3,5-三苄氧基戊烷-1,4-二醇的制备方法,用于杂质标准品的研究。
[0011]
为了实现以上目的,本发明采用以下技术方案:
[0012]
一种瑞德西韦有关物质i,所述有关物质i为(2r,3r,4r)-1,1-二(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-2,3,5-三苄氧基戊烷-1,4-二醇,其结构式为:
[0013]
一种上述瑞德西韦有关物质i的制备方法,包括以下步骤:
[0014]
(1)碘代三嗪(p1)加入有机溶剂中,冷却至-10-30℃,加入三甲基氯硅烷,保温搅拌10-30min;
[0015]
(2)降温至-30-20℃,加入苯基氯化镁,保温搅拌30-60min;
[0016]
(3)加入异丙基氯化镁-氯化锂,保温搅拌30-60min;
[0017]
(4)加入核苷(p2)的有机溶剂溶液,保温搅拌30-60min;
[0018]
(5)加入酸水,有机溶剂萃取,浓缩得到有关物质i粗品,精制得到有关物质i纯品。
[0019]
本发明中所述碘代三嗪(p1)的结构为:所述核苷(p2)的结构为:
[0020]
优选地,所述步骤(1)和步骤(4)中有机溶剂选自乙酸乙酯、乙腈、甲苯、二甲苯、二氯甲烷、四氢呋喃、2-甲基四氢呋喃、甲叔醚、二甲基甲酰胺、氯仿、醋酸异丙酯中的一种或多种;进一步优选为四氢呋喃;所述有机溶剂和碘代三嗪的体积质量比为5-20:1;进一步优选为10-15:1。
[0021]
优选地,步骤(1)中所述三甲基氯硅烷与碘代三嗪的摩尔比为1.0-2.5:1,进一步优选为2.0-2.2:1。反应温度进一步优选为0-5℃。
[0022]
优选地,步骤(2)中所述苯基氯化镁与碘代三嗪的摩尔比为1.5-2.5:1,进一步优选为2.0-2.2:1。反应温度优选为-5-0℃。
[0023]
优选地,步骤(3)中所述异丙基氯化镁-氯化锂与碘代三嗪的摩尔比为0.5-1.2:1,进一步优选为0.9-1.0:1。反应温度优选为-5-0℃。
[0024]
优选地,步骤(4)中所述核苷与碘代三嗪的摩尔比为0.3-1.5:1,进一步优选为0.4-0.6:1。反应温度优选为-5-0℃。
[0025]
优选地,步骤(5)中所述酸选自有机酸、无机酸或其组合;所述有机酸选自甲酸、乙酸、三氟乙酸、甲磺酸、乙磺酸、苯磺酸及对甲苯磺酸中的一种或几种;所述无机酸选自盐酸、硫酸及磷酸中的一种或几种。
[0026]
优选地,步骤(5)中所述有机溶剂选自乙酸乙酯、甲苯、二甲苯、二氯甲烷、甲叔醚、氯仿、醋酸异丙酯中的一种或几种;进一步优选为乙酸乙酯或者二氯甲烷。
[0027]
所述精制具体为:将有关物质i粗品中加入4-10倍质量的溶剂,升温至50-70℃,缓慢降温至0-20℃,过滤得到纯品。
[0028]
优选地,步骤(5)中所述精制过程所用溶剂选自乙酸乙酯、甲苯、二甲苯、甲叔醚、醋酸异丙酯、正己烷、正庚烷、环己烷、石油醚中的一种或几种;进一步优选为甲叔醚。
[0029]
与现有技术相比,本发明具有以下有益效果:
[0030]
1)采用本技术提供的方法制备瑞德西韦有关物质i,瑞德西韦有关物质i的收率可达60-80%之间,收率大幅提高、品质好;相比传统方法,收率可以提高一千倍以上。
[0031]
2)本技术提供的方法,其采用的溶剂可以为四氢呋喃、二氯甲烷、乙酸乙酯、甲叔醚等有机溶剂,未涉及到不常用或昂贵的有机溶剂,在酸性条件下产物手性中心稳定,不会发生异构化。
[0032]
3)在整个制备过程中,所用试剂安全、环保,反应条件温和,无需使用特殊设备及原料,制备周期较短,处理简单,易于操作,成本低。
[0033]
4)本发明得到的有关物质i可作为瑞德西韦、其中间体或其制剂的质量控制中的重要指标,具有制备方法简便、对环境友好的优势。
[0034]
5)本发明通过控制化合物p3中的权利要求1所述杂质的含量小于0.50%,使瑞德西韦中杂质i含量小于0.10%,满足对单一杂质的控制需求。
附图说明
[0035]
图1为通过实施例1过程生产的瑞德西韦原料药的液相检测图;
[0036]
图2为通过实施例1过程生产的瑞德西韦原料药的液质检测图;
[0037]
图3为通过实施例1过程生产的瑞德西韦原料药中有关物质i的液相图谱;
[0038]
图4为通过实施例1过程生产的瑞德西韦原料药中有关物质i的液质图谱;
[0039]
图5为本发明制备的有关物质i的核磁氢谱;
[0040]
图6为本发明制备的有关物质i的碳谱图;
[0041]
图7为本发明制备的瑞德西韦有关物质i编号图;
[0042]
图8为本发明制备的有关物质i的重水交换氢谱;
[0043]
图9为本发明制备的有关物质i重水交换后编号图。
具体实施方式
[0044]
为了使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面结合具体实施例,进一步阐明本发明,但下述实施例仅为本发明的优选实施例,并非全部。基于实施方式中的实施例,本领域技术人员在没有做出创造性劳动的前提下所获得其它实施例,都属于本发明的保护范围。
[0045]
下述实施例中的实验方法,如无特殊说明,均为常规方法,下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。其中本技术具体实施例中所用碘代三嗪(p1)、核苷(p2)、四氢呋喃(thf)、三甲基氯硅烷(tmscl)、苯基氯化镁四氢呋喃溶液(phmgcl)、异丙基氯化镁-氯化锂四氢呋喃溶液(i-prmgcl
·
licl)购自麦克林试剂。下述实施例中所用仪器的生产厂家是台州信立仪器公司,检测仪器的生产厂家为安捷伦公司。
[0046]
实施例1
[0047]
申请人在瑞德西韦原料药的检测中,发现存在一种未知的杂质。
[0048]
生产瑞德西韦原料药的路线图为:
[0049][0050]
通过液相检测图(图1)以及液质检测图(图2)证明上述流程制备得到的产物中包括瑞德西韦。通过液相图谱(图3)和液质图谱(图4)证明制备过程中15min对应的杂质峰为有关物质i,其峰面积比例一般均在0.05-0.10%。经过高分辨质谱及核磁共振表征,该杂质确认为瑞德西韦有关物质i,即(2r,3r,4r)-1,1-二(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-2,3,5-三苄氧基戊烷-1,4-二醇,其结构如下:
[0051][0052]
实施例2
[0053]
一种上述的瑞德西韦有关物质i的制备方法,合成过程表示为:
[0054][0055]
具体包括以下步骤:
[0056]
(1)130g底物p1(0.5mol)溶于1300ml的thf中,降温至5℃。缓慢加入108g的tmscl,保温搅拌0.5h,降温至0℃。
[0057]
(2)缓慢滴加525ml的phmgcl(2m in thf),控制内温-5-0℃,加毕保温搅拌0.5h,冷却至-5℃。(注:phmgcl的摩尔量为0.525*2=1.05mol)
[0058]
(3)滴加330ml的i-prmgcl
·
licl(1.5m in thf),控制内温-5-0℃,加毕保温40min。
[0059]
(4)滴加p2(105g)的thf(200ml)溶液,控制内温-5-0℃,加毕保温1h。
[0060]
(5)滴加稀盐酸淬灭,调节ph至5-6,加入1000ml乙酸乙酯萃取,浓缩有机相,得到粗品;将有关物质i粗品中加入粗品6倍质量的甲叔醚溶剂,升温至60℃保持1-2h,缓慢降温至10℃保持1-2h,过滤得到纯品137g,收率80%,纯度99.2%。
[0061]
实施例3:1.2摩尔比(0.6mol)的苯基氯化镁
[0062]
与实施例1不同仅在于:步骤(2)中phmgcl的加入量为300ml,其他步骤与实验参数均与实施例1相同。精制得到纯品127g,收率74%,纯度98.6%。
[0063]
实施例4:1.2摩尔比(0.6mol)的三甲基氯硅烷
[0064]
与实施例1不同仅在于:步骤(1)中tmscl的加入量为129.6g,其他步骤与实验参数均与实施例1相同。精制得到纯品121g,收率71%,纯度97.8%。
[0065]
实施例5:1.0摩尔比(0.5mol)的核苷
[0066]
与实施例1不同仅在于:步骤(4)滴加209gp2的thf(200ml)溶液,其他步骤与实验参数均与实施例1相同。精制得到纯品111g,收率65%,纯度98.4%。
[0067]
实施例6
[0068]
与实施例1不同仅在于:步骤(2)中温度设置为10℃,其他步骤与实验参数均与实施例1相同。精制得到纯品113g,收率66%,纯度95.8%。
[0069]
对比例1
[0070]
与实施例1不同仅在于:步骤(1)中温度设置为-20℃,其他步骤与实验参数均与实施例1相同。精制得到纯品96g,收率56%,纯度93.5%。
[0071]
对比例2
[0072]
与实施例1不同仅在于:步骤(1)中搅拌时间设置为5min,其他步骤与实验参数均与实施例1相同。精制得到纯品79g,收率46%,纯度92.8%。
[0073]
对比例3
[0074]
与实施例1不同仅在于:步骤(2)中温度设置为30℃,其他步骤与实验参数均与实施例1相同。精制得到纯品84g,收率49%,纯度91.8%。
[0075]
对比例4
[0076]
与实施例1不同仅在于:步骤(2)中搅拌时间设置为20min,其他步骤与实验参数均与实施例1相同。精制得到纯品87g,收率51%,纯度93.6%。
[0077]
对比例5
[0078]
与实施例1不同仅在于:步骤(3)中温度设置为40℃,其他步骤与实验参数均与实施例1相同。精制得到纯品72g,收率42%,纯度92.3%。
[0079]
测试例
[0080]
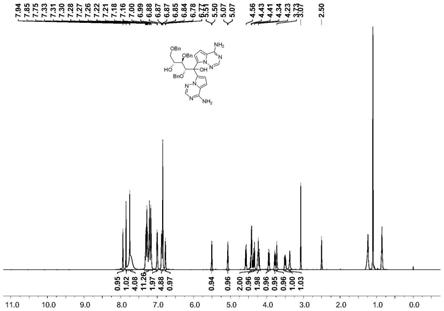
本发明制备的瑞德西韦有关物质i,通过液质和核磁共振结构表征确定合成产物结构正确,对其液质(图4)、核磁共振氢谱(图5)和碳谱(图6)进行了解析。有关物质i的分子式为:c38h38n8o5,分子量计算值为686.30,在图4中阴离子峰为[m-h]:685.10,阳离子峰为[m h]:687.41,与理论计算值相吻合。该化合物编号如图7所示。
[0081]
表1.瑞德西韦有关物质i的核磁解析
[0082]
质子类型化学位移归属h编码质子数o-h3.4ppm,5.0ppmh-9,52hc-h3.5-5.0ppm,5.5ppmh-6,7,8,10,11,12,1311hc-h6.5-7.5ppmh-(ph1,2,3),3,3’,4,4’19hc-h7.7ppm,7.8ppmh-2,2’2hn-h7.7-8.0ppmh-1,1’4h
[0083]
注:重水交换后o-h和n-h化学位移消失,如图8、9所示。
[0084]
表2.瑞德西韦有关物质i的核磁解析
[0085]
质子类型化学位移归属h编码质子数c70-90ppmc-7,8,10,11,12,13,147cc100-120ppmc-3,3’,4,4’,5,5’6cc130-160ppmc-(ph1,2,3),1,1’,2,2’,6,6’24c
[0086]
瑞德西韦有关物质i的用途表征
[0087]
用于瑞德西韦的质量控制
[0088]
通过称量不同质量的p3(纯度大于99.5%,且不包含瑞德西韦杂质i的标准品)和瑞德西韦杂质i(来自实施例2)混合均匀分别得到瑞德西韦杂质i含量为0.10%、0.30%、0.50%、0.70%和0.90%的p3。方法如下:
[0089]
表3.
[0090][0091]
称量后将p3和瑞德西韦杂质i混合均匀,即得到瑞德西韦杂质i含量为0.10%、0.30%、0.50%、0.70%和0.90%的p3。
[0092]
使用瑞德西韦杂质i含量为0.10%的p3制备瑞德西韦的具体实验过程如下:
[0093]
p4的制备:
[0094]
10g p3溶于300g dcm,冷却至-35℃,加入6g的tfoh,10min后加入10g的tmsotf,保温搅拌30min后,冷却至-45℃,加入6g的tmscn,保温搅拌1h,中控检测原料《1%,加入13g的tea淬灭,将反应液缓慢倒入碳酸钠水溶液中,萃取,分液。有机相浓缩,正己烷热打浆粗品,冷却至0-5℃,抽滤。得到8.52g的p4,收率84%。
[0095]
p5的制备:
[0096]
8g p4溶于50g的dcm,冷却至-25℃以下,加入80g的bcl3的dcm溶液(1m),保温搅拌2h,冷却至-25℃以下,加入20g甲醇淬灭,升温至室温,蒸除dcm,碳酸钾水溶液调节ph至6~7左右,打浆,抽滤,烘干得到产品。得产品3.2g,收率80%。
[0097]
p6的制备:
[0098]
3.0g底物加入30ml的丙酮,3.6g甲醇缩丙酮,1g浓硫酸,室温搅拌20min后,升温至65℃搅拌1-2h。降温至0-10℃,加入碳酸钠溶液淬灭反应,蒸除丙酮,加入20g水,20gea,萃取分液,有机相浓缩,得到p6油状物3.2g,收率88%。
[0099]
p8的制备:
[0100]
3.2g p6溶于30ml乙腈,加入4.5g p7,1g无水氯化镁,升温至50℃搅拌30min,加入5g的dipea,保温搅拌1-2h,降温至20℃,用50ml ea稀释,加入适量的5%的柠檬酸水溶液,调节ph至中性,萃取分液,有机相水洗,并用无水硫酸钠干燥,浓缩,得到5.8gp8。收率93%。
[0101]
瑞德西韦的制备:
[0102]
5.8g p8溶于50ml的thf,冷却至0℃,缓慢加入10g浓盐酸,升温至15℃搅拌5h,冷却至0℃,缓慢加入碳酸钾水溶液,调节ph至8左右,50g ea和50g水萃取分液,有机相水洗,浓缩,10g乙醇溶解,滴加40g饮用水析晶,得到瑞德西韦4.6g,收率85%。
[0103]
所制备的瑞德西韦样品经hplc检测,瑞德西韦中杂质i纯度为0.03%。
[0104]
含有0.30%、0.50%、0.70%、0.90%瑞德西韦杂质i的p3分别经上述工艺制备瑞德西韦,瑞德西韦中杂质i的纯度分别为0.06%、0.09%、0.12%、0.15%。
[0105]
p3中该瑞德西韦杂质i不同含量,对通过含有该杂质i的p3制备得到的瑞德西韦结果如下:
[0106]
表4.
[0107][0108][0109]
结论:各大药典有关物质中单一杂质一般要求小于0.10%。当p3中该杂质i的含量为0.50%时,经一系列合成步骤p3中该杂质生成瑞德西韦中杂质i,杂质i为0.09%接近0.10%的指标,严格控制p3中该杂质的含量小于0.50%,能保证瑞德西韦中杂质i符合质量标准的要求。
[0110]
最后应当说明的是,以上内容仅用以说明本发明的技术方案,而非对本发明保护范围的限制,本领域的普通技术人员对本发明的技术方案进行的简单修改或者等同替换,均不脱离本发明技术方案的实质和范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。