1.本发明属于药物化学的技术领域,具体涉及一种富马酸沃诺拉赞中间体及其制备方法,以及用于制备富马酸沃诺拉赞的应用。
背景技术:
2.富马酸沃诺拉赞是由日本武田制药开发的一种新型胃酸分泌抑制剂,于2014年2月向日本提交上市申请。在国内,于2014年5月批准临床。该药物属于一种新型的ppi-钾离子竞争性酸阻滞剂,通过竞争性抑制氢离子/钾离子-atp酶中的钾离子而起作用,是一种可逆的钾离子拮抗剂。与传统的ppi相比,此类药物抑酸效果和质子泵活化情况无关,临床上可明显减少夜间酸突破的产生。可用于治疗非糜烂性胃食管反流病、十二指肠溃疡、胃溃疡、糜烂性食管炎等。富马酸沃诺拉赞结构如下所示:
[0003][0004]
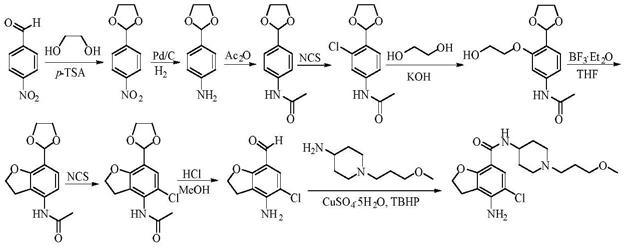
当前关于富马酸沃诺拉赞的制备方法,文献中多有报道。比如专利cn101300229a报道的合成路线如下:
[0005][0006]
此路线工艺步骤较长,处理较为繁琐,同时起始物料1h-吡咯-3-羧酸甲酯不易购买,反应过程中酯基还原为羟基,羟基又氧化为醛基等操作,反应条件较为苛刻,使反应不易控制,且后处理难度大,因此不适合大规模生产。
[0007]
专利cn105085484a报道合成路线如下:
[0008][0009]
同样,该合成路线较长,需经叔丁氧羰基保护胺基后再脱保护,操作步骤繁琐,原子经济性较差,不适合工业化生产。
[0010]
专利cn104860923a报道的合成路线如下:
[0011][0012]
该路线步骤相对简单,但将腈基还原成胺基的过程,需要采用钯、铂、铑等重金属催化氢化,催化剂较昂贵且操作有一定危险性;后续的胺基甲基化利用甲醛类化合物来实现的,甲醛类毒性太大,有引入基因毒性物质的危险,同样不适合进行工业化生产。
[0013]
因此,开发一种新的富马酸沃诺拉赞的制备方法是非常有必要的。
技术实现要素:
[0014]
针对现有文献公开的制备富马酸沃诺拉赞的方法中,合成路线普遍较长,且反应过程多需要氧化还原来实现,操作危险性较大,不利于工业化的实现等问题。本发明提供了一种结构新颖的沃诺拉赞中间体化合物,并且该中间体的制备操作简单。使用该中间体制备富马酸沃诺拉赞,可以避免危险性高的氧化还原操作,避免毒性杂质的引入,既提高了收率,又提高了产品质量,从而克服了现有工艺操作过程复杂的问题,适合工业化生产。
[0015]
本发明提供一种富马酸沃诺拉赞中间体化合物,其具有如式i所示的结构:
[0016][0017]
本发明另一方面,提供一种式i所示中间体化合物的制备方法,具体步骤包括:
[0018][0019]
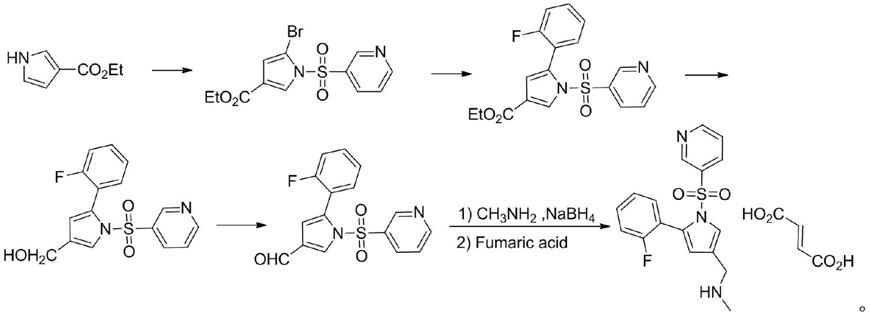
5-(2-氟苯基)-1h-吡咯-3-甲醇和3-吡啶磺酰氯在有机溶剂中反应得到(5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯即式i中间体化合物。
[0020]
优选地,一种式i所示中间体化合物的制备方法,具体步骤包括:
[0021]
将5-(2-氟苯基)-1h-吡咯-3-甲醇、催化剂和敷酸剂加入到有机溶剂中,搅拌降温,然后缓慢滴加3-吡啶磺酰氯溶液,滴加完毕,升温,保温搅拌;加冰水,控制温度,再加酸调节ph,继续搅拌,抽滤得到(5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯。
[0022]
优选地,所述的有机溶剂选自乙腈、四氢呋喃、二氧六环中的一种;优选为乙腈。
[0023]
优选地,所述的5-(2-氟苯基)-1h-吡咯-3-甲醇与有机溶剂的质量体积比为1:6~15,g/ml;优选的质量体积比为1:8~10,g/ml。
[0024]
优选地,所述的5-(2-氟苯基)-1h-吡咯-3-甲醇与3-吡啶磺酰氯的投料摩尔比为1:2~3;优选的摩尔比为1:2.2~2.5。
[0025]
优选地,所述的催化剂为4-二甲氨基吡啶(dmap)或吡啶。
[0026]
优选地,所述的敷酸剂为三乙胺、n-甲基吡咯烷、四甲基乙二胺;优选为三乙胺。
[0027]
优选地,所述的5-(2-氟苯基)-1h-吡咯-3-甲醇与催化剂的摩尔比为1:0.3~1.0;优选的摩尔比为0.5~0.7。
[0028]
优选地,所述的5-(2-氟苯基)-1h-吡咯-3-甲醇与敷酸剂的摩尔比为1:2~3;优选的摩尔比为2.2~2.4。
[0029]
优选地,所述的搅拌降温的温度为-10~10℃;优选的温度为-5~0℃。
[0030]
优选地,所述的升温的温度为10~30℃;优选的温度为10~15℃。
[0031]
优选地,所述的保温搅拌的时间为2~10h;优选的时间为4~6h。
[0032]
优选地,所述的控温温度为0~15℃;优选为5~10℃。
[0033]
优选地,所述的酸为盐酸、硫酸或冰醋酸。
[0034]
优选地,所述的ph为5~7。
[0035]
优选地,继续搅拌的时间为2~4h。
[0036]
本发明还提供一种式i中间体用于制备富马酸沃诺拉赞的用途,具体步骤包括:
[0037][0038]
以(5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯经过胺化得到1-(5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-基)-n-甲基甲胺,然后成盐得到产物富马酸沃诺拉赞。
[0039]
优选地,一种式i中间体用于制备富马酸沃诺拉赞的用途,具体步骤包括:
[0040]
(5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯加入有机溶剂a中,搅拌降温,缓慢加入甲胺试剂,然后升温,保温搅拌反应;反应结束加纯化水,萃取剂萃取,洗涤有机相,浓缩得沃诺拉赞。然后加入有机溶剂b,加热搅拌溶解,加入富马酸,降温继续搅拌反应,反应毕过滤,洗涤,收集固体,干燥得富马酸沃诺拉赞。
[0041]
优选地,所述的有机溶剂a为甲醇、乙酸乙酯、二氯甲烷、氯仿、乙腈;优选为甲醇。
[0042]
优选地,所述的(5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯与有机溶剂a的质量体积比为1∶6~15,g/ml;优选的质量体积比为1:8~10,g/ml。
[0043]
优选地,所述的(5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯与甲胺试剂(以甲胺算)的摩尔比为1∶2~2.6;优选的摩尔比为1:2.1~2.3。
[0044]
优选地,所述的甲胺试剂为甲胺、甲胺甲醇溶液,甲胺水溶液,甲胺乙醇溶液;优选甲胺甲醇溶液。
[0045]
优选地,所述的搅拌降温的温度为-10~10℃;优选的温度为-5~0℃。
[0046]
优选地,所述的升温的温度为25~40℃;优选的温度为35℃。
[0047]
优选地,所述的保温搅拌的时间为5~20h;优选的时间为8~10h。
[0048]
优选地,所述萃取使用的萃取剂为二氯甲烷、氯仿、乙酸乙酯;优选为二氯甲烷。
[0049]
优选地,所述的有机溶剂b为乙酸乙酯、异丙醇、n,n-二甲基甲酰胺;优选为乙酸乙酯。
[0050]
优选地,所述的沃诺拉赞与有机溶剂b的质量体积比为1:5~10,g/ml;优选的质量体积比为1:6~8,g/ml。
[0051]
优选地,所述的加热溶解的温度为40~60℃;优选的温度为50~55℃。
[0052]
优选地,所述的降温继续搅拌的温度为-10℃~10℃,优选的温度为-5℃~0℃。
[0053]
优选地,所述的沃诺拉赞与富马酸的摩尔比为1:1~2;优选的摩尔比为1:1.05~1.2。
[0054]
优选地,所述的搅拌反应的时间为1~4h;优选的时间为2~3h。
[0055]
与现有技术相比,本发明取得的技术效果是:
[0056]
本发明提供了一种新的沃诺拉赞中间体化合物,该化合物可以用于制备富马酸沃诺拉赞,方法简单、反应高效;本发明也提供了一种制备富马酸沃诺拉赞的全新路线,该路
线较现有文献报道的路线反应步骤短,原材料易得,避免了大量有毒有害试剂的使用,具有较高的反应收率及较好的安全性,适合工业化应用。
具体实施方式
[0057]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属本发明要求保护的范围。
[0058]
实施例1式i中间体的制备
[0059]
将5-(2-氟苯基)-1h-吡咯-3-甲醇100g,吡啶20.5g,三乙胺115.7g,加入到800ml乙腈中,搅拌降温到0℃,然后缓慢滴加3-吡啶磺酰氯溶液203.2g,滴加完毕,升温至15℃,保温搅6h后,加冰水200g,控制温度5~10℃,再缓慢滴加0.5mol/l的盐酸,调节ph至6,继续搅拌3h。抽滤收集固体,洗涤,干燥,得到(5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯227.8g。
[0060]
结构确证:esi-ms(m/z):474.05[m h]
;1h nmr(400mhz,dmso-d6)δ:8.86~8.98(m,2h),8.81~8.83(m,1h),8.76~8.79(m,1h),8.65~8.69(d,j=2.0hz,2h),7.79~7.83(m,2h),7.43~7.45(m,1h),7.32~7.36(m,1h),7.11~7.18(m,2h),6.52(s,1h),6.05(s,1h);4.59(s,2h)。
[0061]
实施例2式i中间体的制备
[0062]
将5-(2-氟苯基)-1h-吡咯-3-甲醇100g,吡啶28.8g,三乙胺126.2g,加入到1000ml乙腈中,搅拌降温到-5℃,然后缓慢滴加3-吡啶磺酰氯溶液230.9g,滴加完毕,升温至10℃,保温搅拌6h后,加冰水200g,控制温度5~10℃,再缓慢滴加0.5mol/l的盐酸,调节ph至6,继续搅拌3h。抽滤收集固体,洗涤,干燥,得到(5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯225.8g。esi-ms(m/z):474.05[m h]
。
[0063]
实施例3式i中间体的制备
[0064]
将5-(2-氟苯基)-1h-吡咯-3-甲醇100g,吡啶12.32g,四甲基乙二胺181.3g,加入到600ml二氧六环中,搅拌降温到0℃,然后缓慢滴加3-吡啶磺酰氯溶液263.8g。滴加完毕,升温至30℃,保温搅拌3h后,加冰水300g,5~10℃,再缓慢滴加0.5mol/l的盐酸,调节ph至5,继续搅拌5h。抽滤收集固体,洗涤,干燥,得到(5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯210.5g。esi-ms(m/z):474.05[m h]
。
[0065]
实施例4式i中间体的制备
[0066]
将5-(2-氟苯基)-1h-吡咯-3-甲醇100g,dmap 31.8g,三乙胺105.2g,加入到800ml四氢呋喃中,搅拌降温到0℃,然后缓慢滴加3-吡啶磺酰氯溶液278g,滴加完毕,升温至15℃,保温搅拌10h后,加冰水200g,控制温度5~10℃,再缓慢滴加0.5mol/l的稀硫酸,调节ph至7,继续搅拌2h。抽滤收集固体,洗涤,干燥,得到(5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯188.2g。esi-ms(m/z):474.05[m h]
。
[0067]
实施例5富马酸沃诺拉赞的制备
[0068]
称取(5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯100g,加入到800ml甲醇中,搅拌降温至-5℃,缓慢滴加45.6g 30%的甲胺甲醇溶液;滴加完毕,升温至35℃,保温搅拌10h;反应结束加纯化水400ml,然后用500ml二氯甲烷萃取,食盐
水洗涤150ml
×
2次,无水硫酸钠干燥,过滤,浓缩至干得沃诺拉赞。然后加入乙酸乙酯400ml,加热至50℃搅拌溶解,加入富马酸26g,降温至0℃继续搅拌2h,过滤,乙酸乙酯20ml洗涤,干燥,得白色固体90.5g,hplc:99.72%。esi-ms m/z:462.12[m h]
。
[0069]
实施例6富马酸沃诺拉赞的制备
[0070]
称取(5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-基)-吡啶-3-磺酸甲酯100g,加入到1000ml甲醇中,搅拌降温至0℃,缓慢滴加49.7g 30%的甲胺甲醇溶液。滴加完毕,升温至40℃,保温搅拌8h。反应结束加纯化水300ml,然后用400ml二氯甲烷萃取,食盐水洗涤100ml
×
2次,无水硫酸钠干燥,过滤,浓缩至干得沃诺拉赞。然后加入乙酸乙酯500ml,加热至50℃搅拌溶解,加入富马酸29.2g,降温至30℃继续搅拌4h,过滤,乙酸乙酯30ml洗涤,干燥,得白色固体89.6g,hplc:99.56%。esi-ms m/z:462.12[m h]
。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。