1.本发明属于基因检测技术领域,涉及一种捕获引物及其应用。
背景技术:
2.自然流产(spontaneous abortion,sa)是妊娠早期常见的并发症,发生胚胎停育及自然流产的原因包括遗传学因素、内分泌因素、免疫因素及环境因素等。50%早期自然流产和6%~17%死胎的发生与染色体异常有关,其中染色三体最常见,其次是三倍体、性染色体单体x、四倍体和染色体结构异常。大部分存在染色体异常的胚胎以自然流产的形式终止妊娠,难以存活至出生,而存活到出生的胚胎,多在短期内死亡,或存在多器官系统严重遗传缺陷,严重影响患儿的健康和生活质量。因此,发生自然流产时,检测自然流产妊娠产物(products of conception,poc)的染色体是否异常,对诊断妊娠丢失原因、评估复发风险和指导生育具有重要意义。
3.胚胎植入前遗传学检测(preimplantation genetic testing,pgt)是指在体外受精的胚胎移植前,取胚胎的遗传物质进行分析,诊断是否有异常,筛选健康胚胎移植,防止遗传病传递的方法。目前,pgt过程中常用的技术手段有荧光原位杂交(fish)、snp全基因组微阵列芯片(snp array)和高通量测序(ngs)等。基于高通量测序技术的检测方法是在体外受精后的胚胎发育至3~5天时,取8细胞期的卵裂球细胞或者囊胚期的滋养层细胞,进行单细胞全基因组扩增得到基因组dna,然后构建测序文库进行测序,根据测序结果进行后续分析。
4.目前常用的简化基因组测序是通过限制性内切酶切开基因组dna,对指定部分采用高通量测序,并得到大量遗传多态性标签序列,以将物种全基因组的测序对策充分展现出来的方法。如cn110283892a公开了基于简化基因组测序技术的褐菖鲉基因筛选和挖掘方法,属于分子标记开发技术领域,包括基因组dna提取;基因组dna双酶切;连接反应;pcr扩增;回收序列片段和高通量测序;数据分析筛选和挖掘基因位点。但该方法可减少基因组的复杂度,实施过程简便,节约成本,同时不依靠参考基因组也能得到全基因组中的遗传多态性标签,因此广泛应用于分子标记开发、遗传图谱构建、基因/qtl定位和全基因组关联分析及群体遗传分析与分子育种领域。但现有简化基因组测序可捕获的snp数目少(一般少于20万个),并且其建库流程相对复杂繁琐,或者需要经历末端修复、da尾添加等繁琐建库流程,或者需要使用pippin或胶回收等进行片段分选,同时要求具有更高的实验操作水平以及时间管控能力。综合上述原因,导致简化基因组测序鲜少在辅助生殖领域得以应用和推广。
5.因此,如何提供一种简单、高效且低成本的简化基因组测序方法,是基因检测领域亟需解决的问题之一。
技术实现要素:
6.针对现有技术的不足和实际需求,本发明提供一种捕获引物及其应用,使用所述捕获引物能够通过pcr直接获得可进行上机测序的文库,省去繁琐的打断、末端修复、连接
等建库操作,操作流程简单稳定。
7.为达上述目的,本发明采用以下技术方案:
8.第一方面,本发明提供一种捕获引物,所述捕获引物自5’端起依次包括测序接头、随机序列和酶切位点。
9.本发明中以酶切位点的分析作为核心要素,结合pcr引物易于设计改造和灵活多变的特点,将酶切识别位点引用到pcr引物的3’末端,从而创造出一种不依赖限制性内切酶的简化基因组测序技术,同时,在引物设计时在其5’末端加入相应的测序接头序列,通过简单的pcr扩增步骤即可获得直接能进行二代测序的文库,整个实验流程简单快速易于操作。
10.本发明中,常规测序接头均适用于本发明技术方案,可根据实际需求进行调整。
11.优选地,所述测序接头包括p1接头或a接头。
12.优选地,所述p1接头的核酸序列包括seq id no.1所示的序列。
13.优选地,所述a接头的核酸序列包括seq id no.2所示的序列。
14.seq id no.1:ccactacgcctccgctttcctctctatgggcagtcggtgat。
15.seq id no.2:ccatctcatccctgcgtgtctccgactcag[barcode]gat。
[0016]
优选地,所述随机序列包括4~10个随机碱基,包括但不限于5个、6个、7个、8个或9个。
[0017]
优选地,所述酶切位点包括gtac、catg、ctag、gatc、tcga或acgt中任意一个或至少两个的组合,优选为gtac。
[0018]
本发明中,使用生物信息学手段分析了各种可能的酶切识别位点在人类23对染色体上的分布密度(酶切位点/mb)与均一性,筛选既具有高覆盖均一性又有高密度的位点分布的酶切位点,能够获得密集的片段。
[0019]
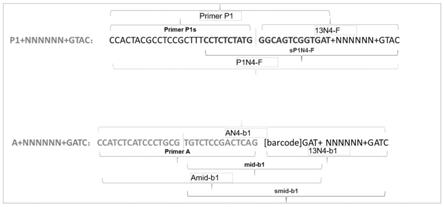
本发明中,将酶切位点碱基序列引入到pcr引物设计中,结合pcr引物易于设计改造和灵活多变的特点,进行了多种可能的设计与组合,以ion torrent测序平台为例,其中p1与a为测序接头序列,将p1序列与a序列直接带入pcr引物设计中,引物形式可以为p1 nnnnnn gtac或a nnnnnn gatc,详细设计原理见图1和图2。
[0020]
优选地,所述捕获引物的核酸序列包括seq id no.3~seq id no.23所示的序列。
[0021]
seq id no.3(p1n4-f):
[0022]
ccactacgcctccgctttcctctctatgggcagtcggtgatnnnnnngtac。
[0023]
seq id no.4(an4-b1):
[0024]
ccatctcatccctgcgtgtctccgactcag[barcode]gatnnnnnngatc。
[0025]
seq id no.5(primer p1s):ccactacgcctccgctttcctctctatg。
[0026]
seq id no.6(primer p1):
[0027]
ccactacgcctccgctttcctctctatgggcagtcggtgat。
[0028]
seq id no.7(primera):ccatctcatccctgcgtgtctccgactcag。
[0029]
seq id no.8(13n4-f):ggcagtcggtgatnnnnnngtac。
[0030]
seq id no.9(13random-f):ggcagtcggtgatnnnnnnmryk。
[0031]
seq id no.10(13n4-b1):[barcode]gatnnnnnngatc。
[0032]
seq id no.11(13random-b1):[barcode]gatnnnnnnkrym。
[0033]
seq id no.12(mid-b1):tgtctccgactcag[barcode]gat。
[0034]
seq id no.13(amid-b1):
[0035]
ccatctcatccctgcgtgtctccgactcag[barcode]gat。
[0036]
seq id no.14(sp1n4-f):
[0037]
cctctctatgggcagtcggtgatnnnnnngtac。
[0038]
seq id no.15(smid-b1):
[0039]
tgtctccgactcag[barcode]gatnnnnnngatc。
[0040]
seq id no.16(capt-catg):gatcgctcttccgnnnnnncatg。
[0041]
seq id no.17(capt-tgac):gatcgctcttccgnnnnnntgac。
[0042]
seq id no.18(capt-agct):gatcgctcttccgnnnnnnagct。
[0043]
seq id no.19(capt-gtca):gatcgctcttccgnnnnnngtca。
[0044]
seq id no.20(capt-ctag):gatcgctcttccgnnnnnnctag。
[0045]
seq id no.21(capt-tcga):gatcgctcttccgnnnnnntcga。
[0046]
seq id no.22(mid-p1):
[0047]
cctctctatgggcagtcggtgatcgctcttccg。
[0048]
seq id no.23(mid-a):
[0049]
ctgcgtgtctccgactcag[barcode]gatcgctcttccg。
[0050]
其中,[barcode]为6~10nt的测序标签序列,用于区分不同样本;nnnnnn,表示4~10个随机简并碱基,m、r、y、k分别为通用简并碱基符号。
[0051]
第二方面,本发明提供第一方面所述的捕获引物在制备简化基因组测序产品中的应用。
[0052]
第三方面,本发明提供一种简化基因组测序试剂盒,所述简化基因组测序试剂盒包括第一方面所述的捕获引物。
[0053]
优选地,所述简化基因组测序试剂盒还包括核酸提取试剂和pcr试剂。
[0054]
第四方面,本发明提供第一方面所述的的捕获引物在简化基因组测序中的应用。
[0055]
第五方面,本发明提供一种构建基因文库的方法,所述构建基因文库的方法包括:
[0056]
使用第一方面所述的捕获引物对样本基因组进行扩增,得到基因文库。
[0057]
第六方面,本发明提供一种简化基因组测序方法,所述简化基因组测序方法包括:
[0058]
使用第一方面所述的捕获引物对样本基因组进行扩增,得到测序文库,对所述测序文库进行测序。
[0059]
优选地,所述扩增包括捕获pcr和特异性pcr。
[0060]
本发明中,针对不同捕获引物的特点,设计了特殊的捕获pcr程序,有目的的特异性扩增预期靶标区域,减少背景噪音干扰,捕获pcr程序设置了一系列由低到高不同的pcr退火温度,以及不同温度下各自不同的退火时长,从而保证捕获扩增的特异性。在捕获pcr程序之后,设计通用特异性pcr扩增程序,进一步降低背景噪音干扰。
[0061]
本发明的简化基因组测序方法使用特殊设计的捕获引物组合和文库构建策略,通过简短的pcr过程即可获得能够进行二代测序的文库,并且所设计的捕获引物组合仅特异性捕获人基因组上部分代表性区域,使用较低的测序数据量即可获得足量高深度的snp信息,满足生殖遗传领域的应用需求,且可省去繁琐的打断、末修、连接等建库操作,操作流程简单稳定,同时对样本起始dna量要求低,最低可对《10ng起始dna获得稳定的检测结果。
[0062]
本发明中,使用所述捕获引物中1对或至少2对以上均能实施本发明简化基因组测序方法。
[0063]
优选地,使用1对引物可以为p1n4-f和an4-b1。
[0064]
优选地,使用2对引物可以为引物对1:primer p1和amid-b1,引物对2:13n4-f和13n4-b1;或者引物对1:primer p1和primer a,引物对2:sp1n4-f和smid-b1;或者引物对1:primer p1s和primer a,引物对2:sp1n4-f和smid-b1;或者引物对1:primer p1和amid-b1,引物对2:sp1n4-f和smid-b1。
[0065]
优选地,使用3对引物可以为引物对1:primer p1和primer a,引物对2:sp1n4-f和mid-b1,引物对3:13n4-f和13n4-b1;或者引物对1:primer p1s和primer a,引物对2:sp1n4-f和mid-b1,引物对3:13n4-f和13n4-b1;或者引物对1:primer p1s和primer a,引物对2:primer p1和mid-b1,引物对3,13n4-f和13n4-b1;或者引物对1:primer p1和amid-b1,引物对2:sp1n4-f和mid-b1,引物对3:13n4-f和13n4-b1;或者引物对1:primer p1s和amid-b1,引物对2:sp1n4-f和mid-b1,引物对3:13n4-f和13n4-b1;或者引物对1,primer p1s和amid-b1,引物对2:primer p1和mid-b1,引物对3:13n4-f和13n4-b1。
[0066]
优选地,使用引物可以为引物对1:primer p1和primer a,引物对2:mid-p1和mid-a,引物对3和capt primer mix,所述capt primer mix由capt-catg、capt-tgac、capt-agct、capt-gtca、capt-ctag或capt-tcga中的2~6种引物混合而成,混合的方法可以是按摩尔数等比例混合,也可以某部分引物更多(如摩尔比为1~3:1)。
[0067]
优选地,所述捕获pcr的程序如表1所示,所述特异性pcr的程序如表2所示。
[0068]
表1
[0069][0070]
表2
[0071][0072]
本发明中,现有测序技术均适用于本发明技术方案,可根据实际需求选择。
[0073]
第七方面,本发明提供一种遗传分析方法,所述遗传分析方法包括对第六方面所述的简化基因组测序方法得到的测序数据进行遗传分析。
[0074]
优选地,所述遗传分析包括流产组织遗传学分析、单基因遗传病检测或染色体结构异常检测。
[0075]
与现有技术相比,本发明具有以下有益效果:
[0076]
(1)本发明特殊设计的捕获引物组合,高密度均一性覆盖人全基因组,能够获得足量(》100万)进行全基因组分型的snp位点,捕获引物上带有测序接头引物序列,简短的pcr过程直接获得可进行上机测序的文库,省去繁琐的打断、末修、连接等建库操作,操作流程简单稳定;
[0077]
(2)本发明建库流程主要为pcr步骤,对样本起始dna量要求低,最低可对《10ng起始dna获得稳定的检测结果,可广泛应用于生殖遗传领域,包括但不限于流产组织遗传学分析、单基因遗传病检测或染色体结构异常检测等。
附图说明
[0078]
图1为捕获引物设计与组合方式示例1图;
[0079]
图2为捕获引物设计与组合方式示例2图;
[0080]
图3为酶切识别位点gtac在人全基因组上的分布图;
[0081]
图4为酶切识别位点gcatgc在人全基因组上的分布图;
[0082]
图5为10ng起始基因组dna量cnv检测结果图;
[0083]
图6为5ng起始基因组dna量cnv检测结果图;
[0084]
图7为1ng起始基因组dna量cnv检测结果图;
[0085]
图8为500pg起始基因组dna量cnv检测结果图;
[0086]
图9为100pg起始基因组dna量cnv检测结果图;
[0087]
图10为三倍体(69xxx)样本cnv检测结果图;
[0088]
图11为三倍体(69xxy)样本cnv检测结果图;
[0089]
图12为基因芯片法检测杂合性丢失(loh)的流产组织样本结果图;
[0090]
图13为本技术检测loh样本的cnv结果图;
[0091]
图14为本技术检测loh样本的cnv局部结果图;
[0092]
图15为各胚胎4号染色体平衡易位分析结果图。
具体实施方式
[0093]
为进一步阐述本发明所采取的技术手段及其效果,以下结合实施例和附图对本发明作进一步地说明。可以理解的是,此处所描述的具体实施方式仅仅用于解释本发明,而非对本发明的限定。
[0094]
实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购获得的常规产品。
[0095]
实施例1
[0096]
本实施例利用生物信息学手段分析了多种可能的酶切识别位点在人类23对染色体上的分布密度(酶切位点/mb)与均一性,结果如表3所示。
[0097]
表3
[0098]
序号酶切识别位点mapd酶切位点/mb1aagctt0.07692772aatt0.089669993acgt0.06727144catg0.043525805cccggg0.34481216ccgc0.261113497ccgg0.28327458ctag0.056323159gaattc0.079426810gatc0.0285444611gcatgc0.115467712gcgc0.287356013gtac0.0342264314raatty0.089297715rcatgy0.047107716rccggy0.268915517rgatcy0.0608142518rgcgcy0.276426819tcga0.1136151420ttaa0.09618421yggccr0.1788668
[0099]
如表示3所示,mapd为评价临近目标间的波动差异,值越小表明均一性越好;酶切位点/mb,指平均每1mb内的酶切位点数,从酶切识别位点的生信分析中,可知gtac、catg、ctag、gatc、tcga和acgt更符合人全基因组遗传标记的分析需求,既具有高覆盖均一性,又具有高密度的位点分布,如图3和图4展示了gtac与gcatgc在人全基因组上的分布对比,可以明显看到gtac分布更密集和均一。
[0100]
实施例2
[0101]
本实施例进行简化基因组测序。
[0102]
胚胎样本采用qiagen repli-g single cell kit进行全基因组扩增,然后使用100ng起始dna进行简化基因组测序,以二步pcr操作为例:
[0103]
step 1,捕获pcr
[0104]
按表4配制反应体系;
[0105]
表4
[0106][0107]
反应体系配制好后,涡旋混匀,短暂离心,然后放置pcr扩增仪上,按表5所示pcr程序进行扩增;
[0108]
表5
[0109][0110][0111]
反应结束后,加入30μl ampure xp磁珠,混匀后室温放置5分钟,放置到磁力架上,等液体清亮,去上清,用200μl的80%酒精清洗2次,25℃干燥后用20μl纯化水洗脱dna;
[0112]
step 2,特异性pcr
[0113]
按表6配制反应体系;
[0114]
表6
[0115]
组分体积(μl)捕获pcr洗脱产物20
pcr酶缓冲液(q5)23mid-p1和primer p1(10μm)1.5mid-a1-4和primer a(10μm)1.5
[0116]
反应体系配制好后,涡旋混匀,短暂离心,然后放置pcr扩增仪上,按表7所示pcr程序进行扩增;
[0117]
表7
[0118][0119]
反应结束后,加入30μl ampure xp磁珠,混匀后室温放置5分钟,放置到磁力架上,等液体清亮,上清液转移到新的离心管中,加入20μl ampure xp磁珠,室温放置5分钟,放置到磁力架上,等液体清亮,去上清,用200μl的80%酒精清洗2次,室温干燥后用20μl纯化水洗脱dna;
[0120]
step3,定量与上机
[0121]
文库定量:吸取2μl以qubit进行浓度测定;
[0122]
上机测序:利用二代测序平台进行ngs测序。
[0123]
实施例3
[0124]
本实施例进行不同起始量细胞系gdna的cnv变异检测。
[0125]
使用已知核型细胞系基因组dna作为样本,gm04126,46,xn;4p16.3-p15.1(del,29.0mb)。分别以10ng、5ng、1ng、500pg和100pg为起始量,使用实施例2所示简化基因组测序方法进行测序并进行cnv变异检测,检测结果如图5~图9所示,起始dna量≥5ng均有稳定的检测结果,1ng以下检测结果开始出现不稳定,其中最低dna起始量为100pg,表明本发明的简化基因组测序方法对样本起始dna量要求低,起始dna量≥5ng均能够获得稳定的检测结果。
[0126]
实施例4
[0127]
本实施例进行流产组织拷贝数异常检测。
[0128]
收集胎儿流产组织,其中三倍体样本通过str技术(对照方法)验证,loh样本经过基因芯片技术(对照方法)验证(图12)。然后使用实施例2所示简化基因组测序方法进行测序并进行分析,三倍体分析结果如图10、图11所示,loh分析结果如图13、图14所示,均包括等位基因频率(af)图和拷贝数值(copy nmuber)图,拷贝数的判断需要综合这2部分进行,如图11中三倍体(69xxy)的copy nmuber图显示拷贝数为2,但af图显示其为三倍体(观察散点的分布范围);图14中copy nmuber图显示拷贝数为2,但af图显示该区域为杂合性缺失(虚线间无散点,表示无杂合位点),从而说明本技术可以实现对三倍体和loh的检测。
[0129]
实施例5
[0130]
本实施例进行胚胎植入前平衡易位家系样本检测,对比pgh结果包括罗氏易位、相互易位和倒位等。
[0131]
募集1个接受辅助生殖的染色体平衡易位携带者家庭(已有基因芯片检测结果),抽取平衡易位携带者夫妻双方外周血样本各5ml于edta抗凝采血管中保存,及他们6枚胚胎活检样本经过全基因组扩增后的产物。其中外周血样本使用核酸提取试剂进行dna抽提,然后对家系与胚胎样本统一使用实施例2所示简化基因组测序方法进行测序并进行胚胎植入前染色体平衡易位分析,结果如表8和图15所示。
[0132]
表8
[0133][0134][0135]
本家系中女方核型为46,xx,t(4;10)(q21;p11.2),男方为正常,子代胚胎363-3为不平衡胚胎,4号染色体发生了缺失,10号染色体发生了重复,说明胚胎363-3遗传了母亲中发生易位的4号染色体,遗传了母亲中正常的10号染色体;故以胚胎363-3为参考时,观察母亲4号单体型遗传情况,具有和其一样单体型的胚胎即为平衡易位携带胚胎(361-4胚胎、361-6胚胎、361-10胚胎、361-12胚胎),具有不一样单体型的胚胎即为正常型胚胎(361-11胚胎),各胚胎4号染色体平衡易位分析结果如图15所示,图15中方框为易位观察区,以不平衡胚胎363-3(胚胎1)为参考样本,图谱中单体型分析从左至右依次为:男方、女方、363-3胚胎(参考)、361-4胚胎、361-6胚胎、361-10胚胎、361-11胚胎、361-12胚胎。
[0136]
实施例6
[0137]
本实施例将本发明方法检测结果与基因芯片检测结果进行对比。
[0138]
本发明测试中,90%左右的read在第10到第20个碱基中包括gtac/tgac/agct,如表9所示,说明本发明构建的文库绝大部分为目标靶序列。
[0139]
表9
[0140][0141][0142]
将本发明检测各染色体信息snp数据和基因芯片芯片检测各染色体信息snp数据比较,lq平衡易位家系5个胚胎各染色体有效位点与芯片进行比较,如表10所示,说明本发明有效信息snp位点数据(胚胎样本)比芯片多。
[0143]
表10
[0144][0145][0146]
综合上述,本发明的简化基因组测序方法可有效应用于生殖遗传领域,包括但不限于流产组织遗传学分析、单基因遗传病检测或染色体结构异常检测等。
[0147]
综上所述,本发明提供了捕获引物及一种简单、高效的简化基因组测序方法,通过简短的pcr过程直接获得可进行上机测序的文库,省去繁琐的打断、末修、连接等建库操作,操作流程简单稳定,且对样本起始dna量要求低,对于遗传分析领域具有重要意义。
[0148]
申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
