1.本发明属于生物信息技术领域,具体涉及一种在全基因组水平上预测植物生长发育转录因子调控的下游靶基因的方法。
背景技术:
2.真核生物转录起始过程十分复杂,往往需要多种蛋白因子协助,转录因子与rna聚合酶ⅱ形成转录起始复合体,能够与基因启动子区域的顺式作用元件发生互作的dna结合蛋白,共同参与转录起始过程。转录因子(tf)广泛存在于真核生物基因组中,并且是生物中最重要的调控因子,在真核生物基因组基因中约有6%是tf基因,虽然比例不高但几乎参与全部基因的转录调控过程。tf由多个基因家族组成,其中在植物中,目前研究较为广泛的有myb、nac、wrky、arf和grf等转录因子。
3.根据tf的作用特点可分为二类;第一类为普遍tf,它们与rna聚合酶ⅱ共同组成转录起始复合体时,转录才能在正确的位置启始。第二类转录因子为组织细胞特异性转录因子,这些tf在特异的组织、细胞或是受到一些类固醇激素\生长因子或其它刺激后,开始表达某些特异蛋白质分子时,才需要该类转录因子。这两类转录因子目前发现都需要和基因的启动子结合才能发挥作用。当前的研究表明,转录因子的表达调控不仅在植物生长发育和细胞的形态等生理活动中发挥重要的调控作用,而且还与植物次生代谢调控和抗逆过程密切相关。因此,对转录因子的表达调控研究具有重要且广泛的意义。并且近些年来研究发现,在重要的经济作物和林木中,转录因子调控机制的研究对于优良品种的选育工作有着重要的指导作用。
4.现阶段的植物转录因子下游调控靶基因的筛选预测方法主要基于转录因子的结合特征,利用生物信息学软件,对结合位点进行预测,将符合条件的候选基础序列(motif)与已经通过实验验证的同源结合位点进行分析比较,最终确定该物种调控的启动子motif序列,并通过结合位点预测下游调控基因。但是现阶段的预测方法存在效率低下的问题。
技术实现要素:
5.有鉴于此,本发明的目的在于提供一种在全基因组水平上预测植物生长发育转录因子调控的下游靶基因的方法,本发明的方法能够快速预测出靶基因。
6.本发明提供了一种在全基因组水平上预测植物生长发育转录因子调控的下游靶基因的方法,包括以下步骤:
7.1)将携带目标转录因子的植物材料作为处理样品,将不携带目标样品的植物材料作为对照样品;分别提取所述处理样品和对照样品的植物基因组dna,打断所述植物基因组dna,得到dna片段,以所述dna片段分别建立处理样品和对照样品的dna文库;
8.2)构建目标转录因子的dap-seq体外蛋白表达载体,得到重组质粒,表达所述重组质粒,得到蛋白表达溶液;
9.3)将所述蛋白表达溶液和蛋白质融合标签第一混合后,再分别与处理样品和对照样品的dna文库第二混合,得到处理样品溶液和对照样品溶液,筛选所述处理样品溶液和对照样品溶液中与重组蛋白结合的dna序列进行测序,分别得到处理样品和对照样品的测序结果;
10.4)分别对所述处理样品和对照样品的测序结果进行预处理,分别得到处理样品和对照样品的待分析数据;所述预处理包括进行质控,过滤掉低质量的序列;
11.5)将所述处理样品和对照样品的待分析数据分别与参考基因组进行比对,分别得到处理样品和对照样品的bam文件;
12.6)对所述处理样品和对照样品的bam文件进行peak-calling分析,得到目标转录因子在全基因组范围内富集的位置信息;
13.7)根据所述目标转录因子在全基因组范围内富集的位置信息,确定待分析序列,对所述待分析序列进行homer-motifspeaks分析,筛选得到目标转录因子调控的基础序列;
14.8)根据所述目标转录因子在全基因组范围内富集的位置信息,利用igv对目标转录因子的结合峰进行可视化分析;
15.9)对步骤7)得到的目标转录因子调控的基础序列进行全基因组基因的启动子比对,比对得到调控下游关键靶基因;
16.步骤1)和步骤2)之间没有时间顺序限制;
17.所述步骤7)和步骤8)之间没有时间顺序限制。
18.优选的,步骤1)中,打断所述植物基因组dna至片段大小为250bp。
19.优选的,步骤1)中,所述植物材料为无性系植物材料。
20.优选的,步骤5)中,利用bowtie2将所述处理样品和对照样品的待分析数据分别与参考基因组进行比对。
21.优选的,步骤6)中,采用macs2.0软件对处理样品和对照样品的bam文件进行peak-calling分析。
22.优选的,步骤6)中,所述peak-calling分析包括:去除处理样品和对照样品中重叠的片段,保留差异片段,所述差异片段的位置信息为目标转录因子在全基因组范围内富集的位置信息。
23.优选的,步骤7)中,所述目标转录因子调控的基础序列的大小为20bp或30bp。
24.优选的,在步骤7)筛选得到目标转录因子调控的基础序列后,还包括以筛选得到的转录因子调控的基础序列作为候选基础序列元件,对所述候选基础序列元件进行分析,过滤假阳性结果,得到校正后的下游调控基础序列元件;所述分析的方法包括:多数据富集分析和多序列比对分析。
25.优选的,本发明在得到校正后的下游调控motif元件后,还包括以包含所述motif元件的启动子基因作为下游调控基因,将所述下游调控基因归类为不同的家族和进行go富集分析。
26.本发明提供了一种在全基因组水平上预测植物生长发育转录因子调控的下游靶基因的方法,本发明的方法基于植物转录因子的dap-seq测序数据,利用macs2.0软件在全基因组中检测调控的启动子的基础序列(motif)元件和位置,并以此为基础,预测转录因子调控的下游靶基因。本发明联合测序数据和生物信息学工具,大大增加了预测的精确度和
通量,进而可在大数据量的基础上快速预测出靶基因,提高预测效率。并且本发明的方法普遍适用于植物学领域。
27.此外,现阶段的植物转录因子下游调控靶基因的筛选预测方法是利用免疫共沉淀技术挖掘转录因子的调控的靶基因,但是该技术需要商业化的抗体,本发明的方法可以不依赖商业化的抗体,能够提高预测和研究的效率。
附图说明
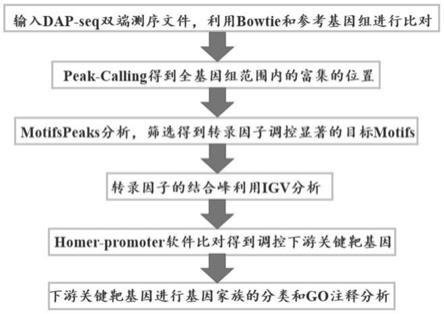
28.图1为本发明实施例中在全基因组水平上预测植物转录因子靶基因的方法的流程图;
29.图2为双端测序的结构示意图;横坐标:0点代表的转录起始位点,纵坐标:富集片段在总片段的百分比;
30.图3表示转录因子调控的启动子motif区域的位置分布;
31.图4表示杨树转录因子调控的启动子motif,其中a为p值最高的两个motif,b为该motif的位点的分布和具体位置;
32.图5表示在杨树中dap-seq的调控位点染色体分布情况;
33.图6表示杨树转录因子调控的下游靶基因go注释分析。
具体实施方式
34.本发明提供了一种在全基因组水平上预测植物生长发育转录因子调控的下游靶基因的方法,包括以下步骤:
35.1)将携带目标转录因子的植物材料作为处理样品,将不携带目标样品的植物材料作为对照样品;分别提取所述处理样品和对照样品的植物基因组dna,打断所述植物基因组dna,得到dna片段,以所述dna片段分别建立处理样品和对照样品的dna文库;
36.2)构建目标转录因子的dap-seq体外蛋白表达载体,得到重组质粒,表达所述重组质粒,得到蛋白表达溶液;
37.3)将所述蛋白表达溶液和蛋白质融合标签第一混合后,再分别与处理样品和对照样品的dna文库第二混合,得到处理样品溶液和对照样品溶液,筛选所述处理样品溶液和对照样品溶液中与重组蛋白结合的dna序列进行测序,分别得到处理样品和对照样品的测序结果;
38.4)分别对所述处理样品和对照样品的测序结果进行预处理,分别得到处理样品和对照样品的待分析数据;所述预处理包括进行质控,过滤掉低质量的序列;
39.5)将所述处理样品和对照样品的待分析数据分别与参考基因组进行比对,分别得到处理样品和对照样品的bam文件;
40.6)对所述处理样品和对照样品的bam文件进行peak-calling分析,得到目标转录因子在全基因组范围内富集的位置信息;
41.7)根据所述目标转录因子在全基因组范围内富集的位置信息,确定待分析序列,对所述待分析序列进行homer-motifspeaks分析,筛选得到目标转录因子调控的motifs;
42.8)根据所述目标转录因子在全基因组范围内富集的位置信息,利用igv对目标转录因子的结合峰进行可视化分析;
43.9)对步骤7)得到的目标转录因子调控的目标motifs进行全基因组基因的启动子比对,比对得到调控下游关键靶基因;
44.步骤1)和步骤2)之间没有时间顺序限制;
45.所述步骤7)和步骤8)之间没有时间顺序限制。
46.本发明首先将携带目标转录因子的植物材料作为处理样品,将不携带目标样品的植物材料作为对照样品;分别提取所述处理样品和对照样品的植物基因组dna,打断所述植物基因组dna,得到dna片段,以所述dna片段分别建立处理样品和对照样品的dna文库。
47.在本发明中,所述植物材料优选为为同一基因型扩繁后的无性系植物材料,保证实验样本一致,所述无性系植物材料优选的通过组织培养技术获得。
48.在本发明中,提取所述处理样品和对照样品的植物基因组dna的方法优选为ctab法。
49.在本发明中,打断所述植物基因组dna至片段大小为250bp,打断所述植物基因组dna采用的仪器优选为bioruptorplus。
50.在本发明中,以所述dna片段分别建立处理样品和对照样品的dna文库的方法优选的包括利用建库试剂盒进行;所述建库试剂盒优选为nextflex-rapid dna-seq kit;所述建库试剂盒完成end repair以及加a过程。
51.本发明构建目标转录因子的dap-seq体外蛋白表达载体,得到重组质粒,表达所述重组质粒,得到蛋白表达溶液。
52.在本发明中,构建目标转录因子的dap-seq体外蛋白表达载体优选的包括以下步骤:将目标转录因子的cds序列重组到pfn9kt7sp6载体,菌落pcr后对构建好的表达载体进行测序,验证构建好的载体是否已经成功连接,确认是否有片段缺失。
53.在本发明中,表达所述重组质粒的方法优选为采用tnt@sp6 high.yieldwheat germ protein expression system(l3260)试剂盒进行重组质粒的表达;所述tnt@sp6 high.yieldwheat germ protein expression system(l3260)试剂盒购自于promega公司。
54.在得到处理样品和对照样品的dna文库以及蛋白表达溶液后,本发明将所述蛋白表达溶液和蛋白质融合标签第一混合后,再分别与处理样品和对照样品的dna文库第二混合,得到处理样品溶液和对照样品溶液,筛选所述处理样品溶液和对照样品溶液中与重组蛋白结合的dna序列进行测序,分别得到处理样品和对照样品的测序结果。
55.在本发明中,所述蛋白融合标签兼具蛋白标记示踪、融合表达和蛋白分离作用。
56.在本发明中,所述第一混合的方式优选为震荡混合;所述第一混合的时间优选为1h;所述第二混合的方式优选为震荡混合;所述第二混合的时间优选为1h;所述第一混合和第二混合分别优选于离心管中进行,在所述第二混合后,优选的还包括在所述第二混合后的体系中加入eb缓冲液,进行震荡;所述震荡的温度优选为98℃,所述震荡的时间为10min,所述震荡的频率为1000rpm;所述震荡后,还包括将震荡后的离心管置于磁力架上至溶液澄清;在所述溶液澄清后取上清液;所述取上清液的时间优选为溶液澄清后1min;所述上清液优选的转移至新离心管;所述上清液的保存温度优选为-20℃。本发明在取上清液后,优选的还包括向含有halotag-beads的离心管中依次加入ddh2o和5
×
sds-page上样缓冲溶液后对离心管进行加热;以所述离心管的规格为1.5ml计,所述ddh2o的加入量优选为每管加入40μl,所述sds-page上样缓冲溶液的加入量优选为每管加入10μl,所述加热的温度优选为
100℃,所述加热的时间优选为10min。
57.在本发明中,所述测序优选的包括以下步骤:
58.利用pcr扩增技术将index加入到与转录因子蛋白结合的dna片段上;利用磁珠筛选加上index的dna片段;利用kapa文库定量试剂盒检测dna文库质量是否合格,构建处理样品和对照样品的链特异性测序文库;利用illumina hiseq2500对所述处理样品和对照样品的链特异性测序文库进行高通量测序获得处理样品和对照样品的原始测序数据。
59.得到处理样品和对照样品的测序结果后,本发明分别对所述处理样品和对照样品的测序结果进行预处理,分别得到处理样品和对照样品的待分析数据;所述预处理包括进行质控,过滤掉低质量的序列;所述质控优选的采用fastqc软件进行,采用fastqc软件的默认参数即可。
60.得到处理样品和对照样品的待分析数据后,本发明将所述处理样品和对照样品的待分析数据分别与参考基因组进行比对,分别得到处理样品和对照样品的bam文件。
61.在本发明中,利用bowtie2将所述处理样品和对照样品的待分析数据分别与参考基因组进行比对;所述bowtie2中设置的参数为默认参数;所述参考基因组为待测物种的已知基因组。
62.得到处理样品和对照样品的bam文件后,本发明对所述处理样品和对照样品的bam文件进行peak-calling分析,得到目标转录因子在全基因组范围内富集的位置信息。
63.在本发明中,优选的采用macs2.0软件对处理样品和对照样品的bam文件进行peak-calling分析。
64.在本发明中,所述peak-calling分析优选的包括:去除处理样品和对照样品中重叠的片段,保留差异片段,所述差异片段的位置信息为目标转录因子在全基因组范围内富集的位置信息。
65.得到目标转录因子在全基因组范围内富集的位置信息后,本发明根据所述目标转录因子在全基因组范围内富集的位置信息,确定待分析序列,对所述待分析序列进行homer-motifspeaks分析,筛选得到目标转录因子调控的motifs。在本发明中,所述目标转录因子调控的motifs的大小优选为20bp或30bp。本发明通过筛选目标转录因子调控的motifs,旨在获得该motifs结合的区域,通过判断该区域是否在启动子区域可以预测下游的功能基因,进而分析转录因子的功能。
66.得到目标转录因子调控的motifs后,本发明优选的还包括以筛选得到的目标转录因子调控的motifs作为候选motif元件,对所述候选motif元件进行分析,过滤假阳性结果,得到校正后的下游调控motif元件;所述分析的方法包括:多数据富集分析和多序列比对分析。
67.本发明在得到校正后的下游调控motif元件后,优选的还包括对包含所述motif元件的启动子基因作为下游调控基因,将所述下游调控基因归类为不同的家族和进行进一步的go富集分析。
68.得到目标转录因子调控的motifs后,本发明对得到的目标转录因子调控的目标motifs进行全基因组基因的启动子比对,比对得到调控下游关键靶基因。本发明对得到的目标转录因子调控的目标motifs进行全基因组基因的启动子比对优选的采用homer-promoter软件进行。
69.得到目标转录因子在全基因组范围内富集的位置信息后,本发明根据所述目标转录因子在全基因组范围内富集的位置信息,利用igv对目标转录因子的结合峰进行可视化分析。
70.本发明中,所述在全基因组水平上预测植物转录因子结合的motif和调控下游靶基因的方法的流程如图1所示。
71.下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。
72.实施例1银腺杨84k的grf15转录因子全基因组范围内调控的下游靶基因的预测
73.下载84k杨基因组的测序文件(从dna research数据库中查找获得:https://academic.oup.com/dnaresearch/article/26/5/423/5580662?searchresult=1#)包括全基因序列文件和gff文件,利用grf15转录因子的dap-seq的测序数据在84k杨全基因组水平上预测该转录因子结合的motif元件,以生信数据库plantpan加以验证,并将结果作为下一步的输入结果。在全基因组水平预测下游调控的靶基因,将获得的下游基因归类为不同的家族成员后,并进一步进行go富集分析。
74.运行步骤:
75.1)通过外源调控因素处理植物材料获得处理样品,未进行外源调控因素处理的样品为对照样品;利用ctab法提取植物基因组dna,构建关键转录因子的dap构建表达载体,对重组质粒进行表达,建库后利用该载体进行按照麦胚蛋白表达系统,利用pcr扩增技术将index加入到与grf蛋白结合的dna片段上,然后利用磁珠进一步筛选加上index的dna片段,再利用kapa文库定量试剂盒检测dna文库质量合格与否,诱导蛋白后获得与重组蛋白结合dna后上机完成测序工作,其中构建该载体的步骤如下:
76.(
ⅰ
)dna片段化及建库
77.利用ctab法提取
78.生长15天84k杨幼嫩的叶片组织的基因组dna,使用bioruptor plus仪器打断至片段大小为250bp。利用磁珠(beckman coulteram-pure xp a63881)筛选目标片段然后利用建库试剂盒(nextflex-rapid dna-seq kit)完成end repair以及加a过程。
79.(
ⅱ
)蛋白表达
80.基于该基因所使用构建表达载体的引物f:5
’‑
cagagcgataacgcgatggagcacgtcatct-caatgga-3’,(seq id no.1);r’:5-agcccgaattcgtttttaata-catgtccatgtgtatggcccca-3’(seq id no.2)将grf的开放阅读框全长重组到pfn9kt7sp6载体,然后,按照叶片蛋白表达系统(promega tnt sp6wheat germmastermix)对grf重组质粒进行表达。
81.(
ⅲ
)dna与蛋白结合以及western-blotting检测
82.将grf的表达蛋白溶液与halotag-beads混合,25℃,1500r/min震荡1h,然后加入dna文库再震荡1h。震荡结束后清洗halotag-beads,向离心管中加入30μl eb溶解,然后再98℃ 1000r/min震荡10min,加热结束后立刻将离心管放在磁力架上,待溶液澄清后2min,将上清转移至新离心管,并保存于-20℃,用于后续实验。然后向含有halotag-beads离心管中加入40μl ddh2o,再加入10μl 5
×
sds-page上样缓冲溶液,然后将离心管放在100℃加热10min。
83.2)提取步骤1)中所述处理样品和对照样品的总dna,利用pcr扩增将index加入到
与grf15蛋白结合的dna片段上,然后利用磁珠进一步筛选除加上index的dna片段构建处理样品和对照样品的链特异性测序文库;用illumina hiseq2500对所述处理样品和对照样品的链特异性测序文库进行高通量测序获得处理样品和对照样品的原始测序数据;
84.3)对于grf15的dap-seq测序的基础结果进行测序结果处理,进行质控,过滤低质量的结果,得到高质量的测序文件;
85.4)过滤所述处理样品和对照样品的原始测序数据获得处理样品和对照样品的cleanreads,参见图2;
86.5)将所述处理样品和对照样品的clean reads分别与84k杨参考基因组比对,利用bowtie2软件将dap-seq测序读长比对到84k基因组(refgen_v3),获得处理样品比对结果与对照样品比对结果;
87.6)对于该测序结果,利用bowtie2将该测序文件包括测序结果和输入文件和所研究物种的基因组建库后进行比对,合成得到bam文件;该步骤具体操作如下:
88.一、将bowtie2处理后的双端测序文件获得的sam文件进行转换并放入后台运行;
89.二、将上一步骤获得的bam文件转换成可以被macs2识别的sorted.bam文件;
90.7)将输入文件和目标序列的bam文件比对后进行macs的peak-calling得到的全基因组范围内的转录因子结合的位置,步骤如下:
91.一利用miniconda下载需要使用的包和软件;包指的是脚本中的模块。
92.二进行peak-calling分析
93.其中设置的参数为:
[0094]-t:实验组,grf转录因子的ip的数据文件;
[0095]
c:gfp的富集结果作为对照组;
[0096]
f:指定输入文件的格式,本次预测使用的bam文件格式;
[0097]
g:杨树的有效基因组大小;
[0098]
其它参数使用默认参数。
[0099]
结果参见图3。
[0100]
对上一步骤所述待分析序列进行homer-motifspeaks分析,筛选得到转录因子调控的目标motifs和位点分布,参见图4和图5。
[0101]
9)转录因子的结合峰利用igv进行可视化分析,转录因子的结合位点主要是在转录起始位点tss附近,所以只有当数据(参考图3,tss 0为转录起始位点,主要柱子都在转录起始位点附近)在tss附近为峰值时数据才正确;
[0102]
10)利用homer-promoter软件对步骤8获得的motif进行全基因组范围内基因的启动子比对分析,得到转录因子调控的下游关键靶基因,参见图6,对于挖掘到的下游关键靶基因进行注释,获得下游关键靶基因参与的调控的富集通路。
[0103]
表1 84k杨全基因组预测出的grf转录因子调控部分靶基因部分结果
[0104][0105]
由以上实施例可知,本发明提供了一种在全基因组水平上预测植物转录因子调控的下游靶基因的一种新方法,该方法能够在大数据量的基础上在全基因范围内快速预测出转录因子调控的motif和下游基因,并且该方法是一种植物普遍适用的方法。
[0106]
尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例而不是全部实施例,人们还可以根据本实施例在不经创造性前提下获得其他实施例,这些实施例都属于本发明保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
