1.本发明属于生物信息学技术领域,具体涉及基于高通量测序寄生虱线粒体基因组组装方法和应用。
背景技术:
2.传统线粒体基因组测序技术主要是基于pcr扩增产物的sanger测序方法,针对已知线粒体基因的突变位点设计引物,进行pcr扩增直接测序。其中通过长pcr产物结合引物步移法(primer walking)是目前小规模线粒体基因组测序的常用方法。
3.线粒体对于大多数真核细胞和生物的生存至关重要。由于这一重要功能,线粒体基因组在深度进化范围内具有非常稳定的结构。因此,线粒体基因组已成为使用频率最高的分子标记之一,被广泛应用于进化、系统发育、种群遗传结构、生物地理学及物种分类鉴定等领域的研究。虱目的线粒体基因组存在高度的重排和裂化,使虱子的线粒体组学研究提高了难度。不利于虱子分子系统学,种群遗传学的研究。
4.寄生虱线粒体基因组裂化是指,线粒体基因组由一个完整的大环裂化为若干个小环。我们在以前的研究中观察到,每个mt微小染色体都有一个不同的编码区,但有一个保守的非编码区。
5.昆虫的传统线粒体基因组测序技术主要是基于pcr扩增产物的sanger测序方法,其中通过长pcr产物结合引物步移法(primer walking)是目前小规模线粒体基因组测序的常用方法。但由于虱目缺乏通用引物信息,使得扩增线粒体基因组部分片段变得困难。而其线粒体基因组中特殊的结构,如重复序列、高a t含量和二级结构也会导致扩增的失败。后随着生物技术的发展,测序成本不断降低以及高通量技术的发展,高通量技术也被广泛应用于虱目的线粒体基因组全序列研究。但是,由于虱目的线粒体基因组的高度重排和裂化,其线粒体的组装和注释成了新的技术难点。
技术实现要素:
6.本发明的目的在,基于高通量法的情况下,提供一种解决虱目线粒体基因组因重排和分裂而难以组装和应用的方法,以解决上述背景技术中虱目的线粒体组装和注释难度大的技术问题。
7.为了解决上述技术问题,本发明提供技术方案包括以下六个步骤:
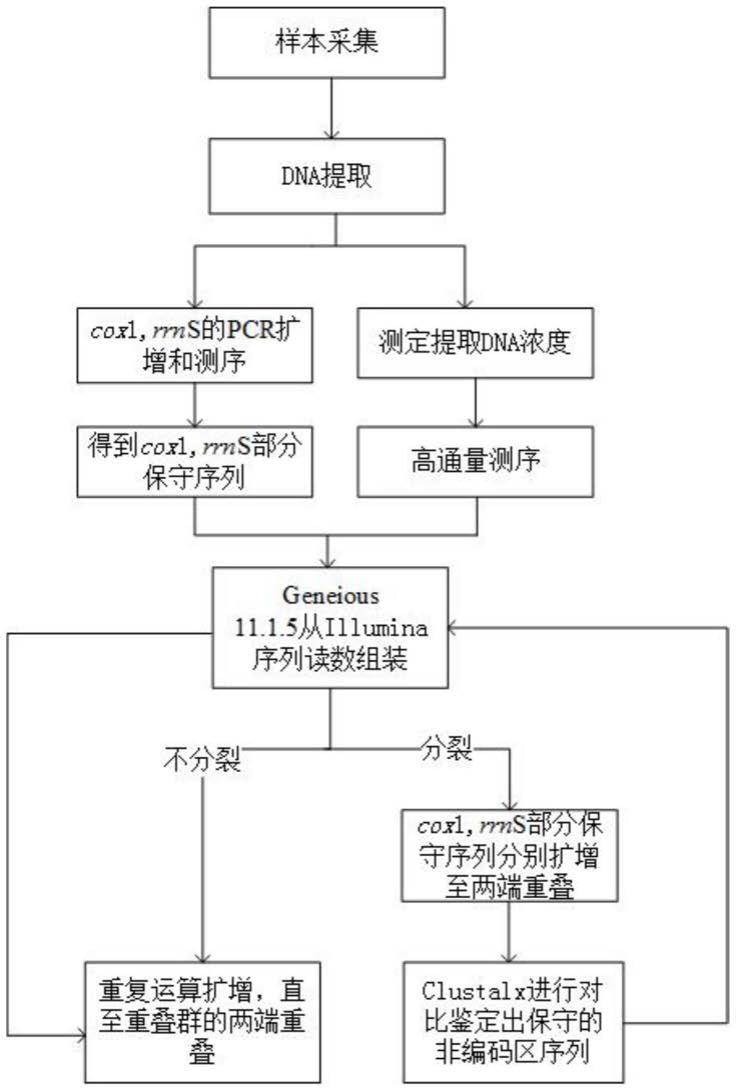
8.(1)根据dnasy组织试剂盒(promega,madison,usa)说明书,从待测寄生虱中提取全基因组dna。
9.(2)利用引物对cox1和rrns基因的扩增和测序。双向测序后分别得到一对正链和负链,将相对应的正链与负链的反向互补链进行对比,得到一段完全重合的序列,此序列即为cox1和rrns相对保守的序列。
10.(3)使用qubit荧光计测定dna浓度。
11.(4)用miseq/hiseq pe300(illumina,san diego,ca,usa)构建成对末端基因组
dna文库(350bp插入),用于高通量测序,收集的原始读数以fastq格式输出。通过删除适配器读取、冗余读取和富含
‘
n’的读取来过滤原始读取;最后,对此寄生虱产生了2gb clean data(256bp的对端读取)。
12.(5)根据cox1和rrns相对保守的序列,使用geneious 11.1.5从illumina序列读数中重新组装重叠群;组装参数为:最小重叠识别率99%,最小重叠150bp。运算至重叠群的两端重叠,表示线粒体基因组排列最终呈一个环形。
13.(6)将两个扩增出来的序列,用软件clustalx进行对比,可得到一段相似或者相同的序列,此序列为保守的非编码区序列。鉴定出保守的非编码区序列,并将其作为比对illumina序列数据集的参考,用上述方法进行运算组装,直至寄生虱线粒体基因组被全部组装完成。基于高通量测序寄生虱线粒体基因组组装的方法的应用,应用为:(1)寄生虱的分子分类与鉴定;(2)物种起源与系统进化。
14.一种基于高通量测序寄生虱线粒体基因组组装的方法的应用,所述应用为:寄生虱的种类鉴定。
15.与现有技术相比,本方案具有以下优点:
16.本方案操作简便,成本低速度快,准确性高。基于高通量法,结合我们之前的研究,根据寄生虱线粒体的特征和规律,即每个mt微小染色体都有一个不同的编码区,但有一个保守的非编码区。据此特征逐一组装虱目的微小线粒体,直到虱目线粒体全基因组被完全组装。组装后的线粒体基因组通过blast进行校准,长pcr法进行验证,完美的解决了寄生虱线粒体基因组的组装与注释的难题。
附图说明
17.图1为本发明线粒体基因组组装的技术流程图。
18.图2为寄生虱线粒体基因组组装注释后第一种结构图。
19.图3为寄生虱线粒体基因组组装注释后第二种结构图。
20.图4为寄生虱线粒体基因组分裂环pcr验证。
21.图5为寄生虱线粒体基因组大环分段pcr验证。
具体实施方式
22.为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
23.实施例1寄生虱线粒体基因组组装(组装流程见图1)
24.一、采集虫体样本,显微镜下观察拍照,并进行初步的形态学鉴定,随后用70%酒精封存虫体,并放入-40℃保存。同时记录样本信息,包括:时间,地点,宿主信息等。
25.二、从酒精中取出待检虫体样本,用双蒸水洗涤虫体3-5遍,使虫体尽可能的少酒精残留。根据dnasy组织试剂盒(promega,madison,usa)说明书,提取待测寄生虱全基因组dna。提取好的dna如近期要用可置于4℃保存,如超过一个星期不用,-20℃保存。
26.三、利用通用引物mtd6(5
′‑
gga gga ttt gga aat tga tta gtt cc-3
′
),mtd11
(5
′‑
act gta aat ata tga tga gct ca-3
′
),对cox1进行pcr扩增和测序;利用引物12sa(5
′‑
tac tat gtt acg act tat-3
′
),12sb(5
′‑
aaa cta gga tta gat acc c-3
′
)对rrns基因pcr扩增和测序。
27.四、根据qubit荧光计的使用说明书,设立标准曲线,选择double-stranded dna(ds dna)模式。测定所提样本的dna浓度。
28.五、用miseq/hiseq pe300(illumina,san diego,ca,usa)构建成对末端基因组dna文库(350bp插入),用于高通量测序,收集的原始读数以fastq格式输出。通过删除适配器读取、冗余读取和富含
‘
n’的读取来过滤原始读数。最后,对此寄生虱产生了2gb clean data(256bp的对端读取)。
29.六、根据cox1和rrns相对保守的序列,使用geneious 11.1.5从illumina序列读数中重新组装重叠群。导入相对保守的序列,与数据库中序列(250bp)进行对比,前后各最小重叠150bp,最小重叠识别率99%,这样,每次运算能够扩增出200bp(前后各100bp)的未知序列,准确性为99%。不断重复运算扩增,直至重叠群的两端重叠,线粒体基因组排列最终呈一个环形。
30.七、将两个扩增出来的序列,用软件clustalx进行对比。鉴定出保守的非编码区序列,并将其作为比对illumina序列数据集的参考,用上述方法进行运算组装,直至寄生虱线粒体基因组被全部组装完成,线粒体基因组结构图见图2和图3。
31.实施例2应用
32.一、对已经组装序列在ncbi上进行blast校准,根据已经组装的序列,用软件premier5.0设计引物,进行pcr验证。pcr产物进行琼脂糖凝胶电泳,根据maker确定每对引物所扩增出的序列长度,与此前被组装的序列长度进行对比验证。验证结果见图4和图5。
33.二、使用同属寄生虱线粒体基因组的全序列,用软件clustalx将其中的基因逐个与新组装序列进行对比,确定序列中基因的大概位置。在ncbi的orf finder上将序列进行扫描,找出蛋白编码的基因的位置。使用arwen和程序trnascan-se,扫描出trna基因的大概位置,并根据trna的二级结构图来手动调整并确定trna基因位置。将以上3项结果分别进行两两验证,确定基因组中全部基因的位置。
34.三、用软件mega 6.0将已经确定蛋白编码基因的核苷酸序列转化fastq格式,再在软件mega 6.0中导入,推导出其氨基酸序列。
35.四、在ncbi数据库中进行blast搜索各个基因的核苷酸序列和氨基酸序列,用以鉴定蛋白质编码基因和rrna基因。
36.五、形态学结合宿主信息对寄生虱进行初步的种类鉴定,利用组装注释好的寄生虱线粒体基因组在ncbi上进行同源性对比,对初步的种类鉴定的结果进行验证。以阐明该寄生虱的分类地位。
37.六、将组装注释好的寄生虱线粒体基因组与ncbi上其他已经解码寄生虱线粒体基因组,构建系统发育树,进行系统发育关系分析,以阐明寄生虱的进化关系。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
