1.本发明涉及有机合成技术领域,尤其是涉及一种氟噻草胺的制备方法。
背景技术:
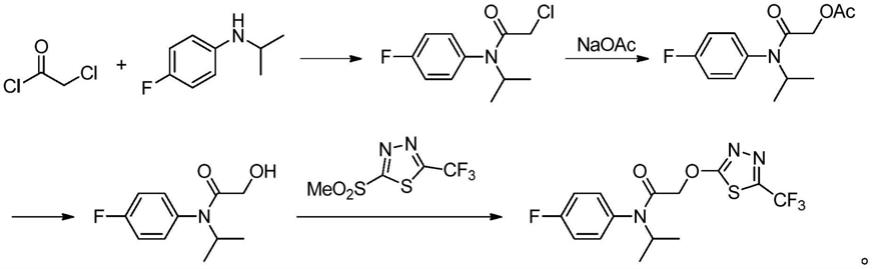
2.氟噻草胺(flufenacet)化学名称为4'-氟-n-异丙基-2-(5-三氟甲基-1,3,4-噻二唑-2-基氧)乙酰基苯胺,4'-fluoro-n-isopropyl-2-(5-trifluoromethyl-1,3,4-thiadiazol-2-yloxy)acetanilide,是一种芳氧酰胺类除草剂,主要通过抑制细胞分裂与生长来发挥作用,具有除草活性高、杀草谱广、适用作物宽、高度安全等优点,可有效防除一年生禾本科杂草及阔叶杂草的芽前、芽后早期除草。最早由拜耳作物科学于1998年推出。在制备过程中,使用2-甲砜基-5-三氟甲基-1,3,4-噻二唑为起始原料,与2-羟基-n-(4-氟苯胺)-n-(1-甲基乙基)乙酰胺反应得到产物氟噻草胺,合成路线如下:
[0003][0004]
上述合成路线的缺点是在中间体2-羟基-n-(4-氟苯胺)-n-(1-甲基乙基)乙酰胺的合成过程中形成大量的含氯废产物。其次,氯乙酰氯是危险品,其运输和存储均受到限制;使用过程中放热快,滴加过快容易升温冲料。另外,上述合成路线步骤较多,其中每增加一个步骤就会导致产率降低,增加纯化次数,造成最后的收率不佳。如何减少甚至不产生含氯废产物,不使用危险物料减少安全风险,减少反应步骤提高收率,是氟噻草胺制备过程中亟待解决的问题。
[0005]
有鉴于此,特提出本发明。
技术实现要素:
[0006]
本发明的目的在于提供氟噻草胺的制备方法,以解决现有技术中存在的原料安全性低、步骤繁琐、收率低等技术问题。
[0007]
为了实现本发明的上述目的,特采用以下技术方案:
[0008]
氟噻草胺的制备方法,包括如下步骤:
[0009]
(a)2-甲砜基-5-三氟甲基-1,3,4-噻二唑或2-氯-5-三氟甲基-1,3,4-噻二唑与化合物a于含碱的溶剂中反应得到化合物b;
[0010]
(b)化合物b与4-氟-n-异丙基苯胺在催化剂的作用下,于溶剂中进行酯交换反应,得到氟噻草胺;
[0011]
其中,化合物a和化合物b的结构式分别如下:
[0012]
r选自碳数为1~4的烷基中的任一种或多种。
[0013]
本发明的氟噻草胺的制备方法,原料易得,且原料安全可靠,同时制备过程中基本不产生含氯废弃物。并且,本发明的氟噻草胺以乙醇酸酯作为起始原料,只需要两步即可得到氟噻草胺产品,大大缩短了反应路线,简化了后处理操作等。
[0014]
在本发明的具体实施方式中,所述r选自甲基(me)、乙基(et)、正丁基(n-bu)和叔丁基(t-bu)中的任一种或多种。
[0015]
在本发明的具体实施方式中,步骤(a)中,所述化合物a与所述2-甲砜基-5-三氟甲基-1,3,4-噻二唑或所述2-氯-5-三氟甲基-1,3,4-噻二唑的摩尔比为1﹕(0.5~2)。
[0016]
在本发明的具体实施方式中,步骤(a)中,所述碱包括氢氧化钠、碳酸钠、碳酸钾和甲醇钠中的任一种或多种。
[0017]
在本发明的具体实施方式中,步骤(a)中,所述反应的温度为-5~40℃。
[0018]
在本发明的具体实施方式中,步骤(a)中,所述溶剂包括水和有机溶剂。进一步的,所述有机溶剂包括甲苯。
[0019]
在本发明的具体实施方式中,步骤(b)中,所述化合物b与所述4-氟-n-异丙基苯胺的摩尔比为1﹕(0.5~2)。
[0020]
在本发明的具体实施方式中,步骤(b)中,所述催化剂包括氢氧化钠、碳酸钠、碳酸钾、甲醇钠、三乙胺、叔丁醇钾、氨气、吡啶、三氟甲磺酸镍和醋酸中的任一种或多种。
[0021]
在本发明的具体实施方式中,步骤(b)中,所述反应的温度为20~100℃。
[0022]
在本发明的具体实施方式中,步骤(b)中,所述溶剂包括水和有机溶剂。进一步的,所述有机溶剂包括甲苯。
[0023]
与现有技术相比,本发明的有益效果为:
[0024]
本发明以乙醇酸酯作为起始原料,只需要两步就可以得到氟噻草胺产品,反应路线短,产品收率高,产品质量好;并且乙醇酸酯作为原料,安全性高且无腐蚀性,制备过程基本不产生含氯废弃物,并且来源广泛。
具体实施方式
[0025]
下面将结合具体实施方式对本发明的技术方案进行清楚、完整地描述,但是本领域技术人员将会理解,下列所描述的实施例是本发明一部分实施例,而不是全部的实施例,仅用于说明本发明,而不应视为限制本发明的范围。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0026]
氟噻草胺的制备方法,包括如下步骤:
[0027]
(a)2-甲砜基-5-三氟甲基-1,3,4-噻二唑或2-氯-5-三氟甲基-1,3,4-噻二唑与化合物a于含碱的溶剂中反应得到化合物b;
[0028]
(b)化合物b与4-氟-n-异丙基苯胺在催化剂的作用下,于溶剂中进行酯交换反应,
得到氟噻草胺;
[0029]
其中,化合物a和化合物b的结构式分别如下:
[0030]
r选自碳数为1~4的烷基中的任一种或多种。
[0031]
本发明的氟噻草胺的制备方法,原料易得,且原料安全可靠,同时制备过程中基本不产生含氯废弃物。并且,本发明的氟噻草胺以乙醇酸酯作为起始原料,只需要两步即可得到氟噻草胺产品,大大缩短了反应路线,简化了后处理操作等。
[0032]
现有技术中以氯乙酰氯作为起始原料,需要四步反应才能得到氟噻草胺产品。并且,氯乙酰氯是危化品,具有酸性腐蚀性,氯乙酰氯的运输过程需要危险品车,存储需要专用储罐,存储及运输安全性低且成本高。氯乙酰氯受热或遇水分解反应,会释放出有毒的腐蚀性烟气,造成安全隐患。本发明采用的乙醇酸酯不是危化品,遇水稳定且无腐蚀性。
[0033]
使用氯乙酰氯,产生氯化钠,只能以固体危废处理,处理费用昂贵。而本发明中使用乙醇酸酯不产生氯化钠,产生的甲醇或者乙醇可以回收利用。
[0034]
使用氯乙酰氯作为起始原料,原料中的杂质二氯乙酰氯含量通常在0.5%左右,会影响制得的氟噻草胺产品的质量。乙醇酸酯中没有相应的杂质,得到的氟噻草胺产品质量好。
[0035]
此外,使用氯乙酰氯作为起始原料制备氟噻草胺的过程中,需要用醋酸钠进行取代反应,然后用氢氧化钠水解,反应操作复杂,增加了物料及溶剂的用量,且均需要升温操作,增加了能耗。
[0036]
在本发明的具体实施方式中,所述r选自甲基(me)、乙基(et)、丙基(n-pr)、异丙基(i-pr)、正丁基(n-bu)、异丁基(i-bu)和叔丁基(t-bu)中的任一种或多种。进一步的,所述r选自甲基(me)、乙基(et)、正丁基(n-bu)和叔丁基(t-bu)中的任一种或多种。
[0037]
当r为甲基时,所述化合物a为乙醇酸甲酯;当r为乙基时,所述化合物a为乙醇酸乙酯;当r为正丁基时,所述化合物a为乙醇酸丁酯;当r为叔丁基时,所述化合物a为乙醇酸叔丁酯。其中,r选自多种基团中的任一种或多种指代,可采用一种或同时采用两种以上化合物a作为原料制备氟噻草胺。
[0038]
在本发明的具体实施方式中,步骤(a)中,所述化合物a与所述2-甲砜基-5-三氟甲基-1,3,4-噻二唑或所述2-氯-5-三氟甲基-1,3,4-噻二唑的摩尔比为1﹕(0.5~2)。
[0039]
如在不同实施方式中,步骤(a)中,所述化合物a与所述2-甲砜基-5-三氟甲基-1,3,4-噻二唑或所述2-氯-5-三氟甲基-1,3,4-噻二唑的摩尔比可以为1﹕0.5、1﹕0.8、1﹕1、1﹕1.2、1﹕1.5、1﹕1.8、1﹕2。
[0040]
在本发明的具体实施方式中,步骤(a)中,所述碱包括氢氧化钠、碳酸钠、碳酸钾和甲醇钠中的任一种或多种。
[0041]
在本发明的具体实施方式中,步骤(a)中,所述碱与所述化合物a的摩尔比为(1~1.5)﹕1。
[0042]
如在不同实施方式中,步骤(a)中,所述碱与所述化合物a的摩尔比可以为1﹕1、1.1﹕1、1.2﹕1、1.3﹕1、1.4﹕1、1.5﹕1等等。在实际操作中,所述碱的用量可根据实际情况进行
调整。
[0043]
在本发明的具体实施方式中,步骤(a)中,所述反应的温度为-5~40℃。
[0044]
如在不同实施方式中,步骤(a)中,所述反应的温度可以为-5℃、0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃等等。
[0045]
在实际操作中,步骤(a)的反应时间可根据实际tlc监测到的反应程度进行调整,可以为1~4h,如2h等等。
[0046]
在本发明的具体实施方式中,步骤(a)中,所述溶剂包括水和有机溶剂。进一步的,所述有机溶剂包括甲苯。
[0047]
如在不同实施方式中,步骤(a)中,所述有机溶剂的用量与所述化合物a的比例可以为400~600ml/1mol,表示相对于每1mol的化合物a,使用的有机溶剂的用量为400~600ml。
[0048]
在本发明的具体实施方式中,步骤(a)包括:2-甲砜基-5-三氟甲基-1,3,4-噻二唑或2-氯-5-三氟甲基-1,3,4-噻二唑与化合物a于有机溶剂中混合,加入碱的水溶液,反应得到含有化合物b的反应液,后处理收集所述化合物b。
[0049]
如在不同实施方式中,步骤(a)中,可预先将碱溶解于水中得到碱的水溶液,然后再加入其余物料(2-甲砜基-5-三氟甲基-1,3,4-噻二唑或所述2-氯-5-三氟甲基-1,3,4-噻二唑、化合物a和有机溶剂)中。所述有机溶剂与所述碱的水溶液的体积比可以为(2~3)﹕1;所述碱的水溶液的质量分数可以为10wt%~40wt%。
[0050]
在本发明的具体实施方式中,所述后处理包括:收集所述反应液中的有机相,然后减压蒸馏除溶得到所述化合物b。进一步的,所述减压蒸馏的温度为70~75℃。
[0051]
在实际操作中,步骤(a)也可先不进行后处理,将反应得到的含有化合物b的反应液直接进行下一步反应制备氟噻草胺。
[0052]
在本发明的具体实施方式中,步骤(b)中,所述化合物b与所述4-氟-n-异丙基苯胺的摩尔比为1﹕(0.5~2)。
[0053]
如在不同实施方式中,步骤(b)中,所述化合物b与所述4-氟-n-异丙基苯胺的摩尔比可以为1﹕0.5、1﹕0.8、1﹕1、1﹕1.2、1﹕1.5、1﹕1.8、1﹕2。
[0054]
在本发明的具体实施方式中,步骤(b)中,所述催化剂包括氢氧化钠、碳酸钠、碳酸钾、甲醇钠、三乙胺、叔丁醇钾、氨气、吡啶、三氟甲磺酸镍、磷酸钠和醋酸中的任一种或多种。
[0055]
在本发明的具体实施方式中,步骤(b)中,所述催化剂与所述化合物b的摩尔比为(0.05~1.5)﹕1。
[0056]
如在不同实施方式中,所述催化剂与所述化合物b的摩尔比可以为0.05﹕1、0.1﹕1、0.5﹕1、1﹕1、1.1﹕1、1.2﹕1、1.3﹕1、1.4﹕1、1.5﹕1等等。在实际操作中,所述催化剂的用量可根据实际情况进行调整,如也可以为0.0005﹕1等等,可催化反应进行即可。
[0057]
在本发明的具体实施方式中,步骤(b)中,所述反应的温度为20~100℃。
[0058]
如在不同实施方式中,步骤(b)中,所述反应的温度可以为20℃、30℃、40℃、50℃、60℃、70℃、80℃、90℃、100℃等等。
[0059]
在实际操作中,步骤(b)的反应时间可根据实际tlc监测到的反应程度进行调整,可以为1~4h,如2h等等。
[0060]
在本发明的具体实施方式中,步骤(b)中,所述溶剂包括水和有机溶剂。进一步的,所述有机溶剂包括甲苯。
[0061]
如在不同实施方式中,步骤(b)中,所述有机溶剂的用量与所述化合物b的比例可以为400~600ml/1mol,表示相对于每1mol的化合物b,使用的有机溶剂的用量为400~600ml。
[0062]
在本发明的具体实施方式中,步骤(b)包括:化合物b与4-氟-n-异丙基苯胺于有机溶剂中混合,加入催化剂,反应得到含有氟噻草胺的反应液,后处理收集所述氟噻草胺。
[0063]
在实际操作中,催化剂可以水溶液的形式加入。
[0064]
如在不同实施方式中,步骤(b)中,可预先将催化剂溶解于水中得到催化剂的水溶液,然后再加入其余物料(化合物b、4-氟-n-异丙基苯胺和有机溶剂)中。所述有机溶剂与所述催化剂的水溶液的体积比可以为(2~3)﹕1;所述催化剂的水溶液的质量分数可以为1wt%~30wt%。
[0065]
在本发明的具体实施方式中,氟噻草胺的制备方法,包括如下步骤:
[0066]
(a)2-甲砜基-5-三氟甲基-1,3,4-噻二唑或2-氯-5-三氟甲基-1,3,4-噻二唑与化合物a于含碱的溶剂中反应得到含有化合物b的反应液;
[0067]
(b)含有化合物b的反应液与4-氟-n-异丙基苯胺混合进行酯交换反应,得到氟噻草胺。
[0068]
本发明的氟噻草胺的制备方法中,在步骤(a)反应结束后,可先不进行后处理,直接在步骤(a)得到的反应液中加入4-氟-n-异丙基苯胺混合反应,得到含氟噻草胺的反应液,分离得到氟噻草胺纯品,简化了后处理操作。
[0069]
在实际操作中,可向步骤(a)得到的反应液中加入催化剂的水溶液及4-氟-n-异丙基苯胺,或者直接增加步骤(a)中的碱及溶剂的用量(增加的碱及溶剂的容量按照步骤(b)所需的量进行调整),反应得到反应液,然后步骤(b)中向反应液中加入4-氟-n-异丙基苯胺进行反应即可。
[0070]
在实际操作中,步骤(a)和步骤(b)可在釜式反应器或微通道反应器中进行。如在微通道反应器中进行,微通道反应器的内径可以为1~50mm,停留时间可以为20~200s,反应液在微通道反应器中的流速可以为1~5m/min。
[0071]
本发明的合成路线可参考如下:
[0072][0073]
实施例1
[0074]
本实施例提供了氟噻草胺的制备方法,合成路线如下:
[0075][0076]
具体的,包括如下步骤:
[0077]
(1)称取乙醇酸甲酯90g(1mol)、2-甲砜基-5-三氟甲基-1,3,4-噻二唑232g(1mol,1.0eq)及甲苯500ml一次性加入1000ml三口瓶中,搅拌混合得到混合溶液;向所述混合溶液中加入20wt%氢氧化钠的水溶液200ml,在20℃条件下搅拌反应2h得到反应液;分离除去所述反应液中的水相,收集有机相,然后将所述有机相于70~75℃减压蒸馏除去甲苯得到2-乙醇酸甲酯-5-三氟甲基-1,3,4-噻二唑(化合物b1)235g,收率为97.0%,hplc检测含量为99.3%。
[0078]
(2)称取步骤(1)中全批产物2-乙醇酸甲酯-5-三氟甲基-1,3,4-噻二唑(235g)、4-氟-n-异丙基苯胺149g(1.0eq)及甲苯(500ml)一次性加入2000ml三口瓶中,搅拌混合得到混合溶液;向所述混合溶液中加入20wt%氢氧化钠水溶液200ml,升温,在50℃的条件下,搅拌反应2h得到反应液;分离除去所述反应液中的水相,收集有机相,然后将所述有机相于70~75℃减压蒸馏除去甲苯收集固体,将固体烘干得到氟噻草胺335g,收率95.1%。hplc检测含量为99.6%。
[0079]
实施例2
[0080]
本实施例参考实施例1的氟噻草胺的制备方法,区别仅在于:步骤(1)中的碱不同。本实施例采用300ml的35.3wt%的碳酸钠的水溶液替换实施例1的步骤(1)中的200ml的20wt%氢氧化钠的水溶液。步骤(1)得到的2-乙醇酸甲酯-5-三氟甲基-1,3,4-噻二唑(化合物b1)的质量为230g,收率为95.0%,hplc检测含量为99.1%。
[0081]
实施例3
[0082]
本实施例参考实施例1的氟噻草胺的制备方法,区别仅在于:步骤(2)中的催化剂不同。本实施例采用1000ml的38wt%的磷酸钠(以磷酸钠十二水合物计)的水溶液替换实施例1的步骤(2)中的200ml的20wt%氢氧化钠的水溶液。步骤(2)得到的氟噻草胺的质量为340g,收率为96.5%,hplc检测含量为99.1%。
[0083]
实施例4
[0084]
本实施例参考实施例1的氟噻草胺的制备方法,区别仅在于:步骤(2)中的催化剂不同。本实施例采用20ml的20wt%的醋酸水溶液替换实施例1的步骤(2)中的200ml的20wt%氢氧化钠的水溶液。步骤(2)得到的氟噻草胺的质量为330g,收率为93.6%,hplc检测含量为99.3%。
[0085]
实施例5
[0086]
本实施例参考实施例1的氟噻草胺的制备方法,区别仅在于:步骤(2)中的催化剂不同。本实施例采用10ml的2wt%的三氟甲磺酸镍水溶液替换实施例1的步骤(2)中的200ml的20wt%氢氧化钠的水溶液。步骤(2)得到的氟噻草胺的质量为331g,收率为93.9%,hplc检测含量为99.0%。
[0087]
实施例6
[0088]
本实施例提供了氟噻草胺的制备方法,包括如下步骤:
[0089]
(1)称取乙醇酸甲酯90g(1mol)、2-甲砜基-5-三氟甲基-1,3,4-噻二唑232g(1mol,1.0eq)及甲苯500ml一次性加入1000ml三口瓶中,搅拌混合得到混合溶液;向所述混合溶液中加入20wt%氢氧化钠的水溶液400ml,在20℃条件下搅拌反应2h得到反应液。
[0090]
(2)向步骤(1)得到的反应液中加入4-氟-n-异丙基苯胺153g(1mol,1.0eq),升温至50℃,搅拌反应2h得到反应液;分离除去所述反应液中的水相,收集有机相,然后将所述有机相于70~75℃减压蒸馏除去甲苯收集固体,将固体烘干得到氟噻草胺359g,总收率98.8%。hplc检测含量为99.1%。
[0091]
实施例7
[0092]
本实施例提供了氟噻草胺的制备方法,包括如下步骤:
[0093]
甲苯500ml、2-甲砜基-5-三氟甲基-1,3,4-噻二唑232g(1mol,1.0eq)混合均匀,与乙醇酸甲酯90g(1mol)、20wt%氢氧化钠水溶液200ml进入混合器,混合均匀后进入内径为10mm的微通道反应器,设定停留时间为30s,温度20℃,反应液在反应器中的流速为3.0m/min。出料口料液与4-氟-n-异丙基苯胺153g(1mol,1.0eq)进入混合器,混合均匀后进入内径为15mm的微通道反应器,设定停留时间为20s,温度50~55℃,反应液在反应器中的流速为5.0m/min。收集出料口料液,分离除去水相,收集有机相,然后将有机相于70~75℃减压蒸馏除去甲苯收集固体,将固体烘干得到氟噻草胺362g,总收率99.6%。hplc含量99.5%。
[0094]
实施例8
[0095]
本实施例提供了氟噻草胺的制备方法,合成路线如下:
[0096][0097]
具体的参考实施例1,区别仅在于:将步骤(1)的乙醇酸甲酯替换为等摩尔的乙醇酸乙酯。
[0098]
步骤(1)得到的2-乙醇酸乙酯-5-三氟甲基-1,3,4-噻二唑(化合物b2)238g,收率为92.9%,hplc检测含量为99.3%;
[0099]
步骤(2)称取步骤(1)中全批产物2-乙醇酸乙酯-5-三氟甲基-1,3,4-噻二唑(238g),得到的氟噻草胺316g,收率为93.6%,hplc检测含量为98.1%。
[0100]
实施例9
[0101]
本实施例提供了氟噻草胺的制备方法,合成路线如下:
[0102][0103]
具体的参考实施例1,区别仅在于:将步骤(1)的乙醇酸甲酯替换为等摩尔的乙醇酸丁酯。
[0104]
步骤(1)得到的2-乙醇酸丁酯-5-三氟甲基-1,3,4-噻二唑(化合物b3)274g,收率为96.4%,hplc检测含量为97.1%;
[0105]
步骤(2)称取步骤(1)中全批产物2-乙醇酸丁酯-5-三氟甲基-1,3,4-噻二唑274g,得到的氟噻草胺310g,收率为88.5%,hplc检测含量为97.3%。
[0106]
实施例10
[0107]
本实施例提供了氟噻草胺的制备方法,合成路线如下:
[0108][0109]
具体的参考实施例1,区别仅在于:将步骤(1)的乙醇酸甲酯替换为等摩尔的乙醇酸叔丁酯。
[0110]
步骤(1)得到的2-乙醇酸叔丁酯-5-三氟甲基-1,3,4-噻二唑(化合物b4)266g,收率为93.6%,hplc检测含量为97.1%;
[0111]
步骤(2)称取步骤(1)中全批产物2-乙醇酸叔丁酯-5-三氟甲基-1,3,4-噻二唑266g,得到的氟噻草胺313g,收率为92.1%,hplc检测含量为96.3%。
[0112]
实施例11
[0113]
本实施例提供了氟噻草胺的制备方法,合成路线如下:
[0114][0115]
具体的,包括如下步骤:
[0116]
(1)称取乙醇酸甲酯90g(1mol)、2-氯-5-三氟甲基-1,3,4-噻二唑188g(1mol,1.0eq)及甲苯500ml一次性加入1000ml三口瓶中,搅拌混合得到混合溶液;向所述混合溶液中加入20wt%氢氧化钠的水溶液200ml,在20℃条件下搅拌反应2h得到反应液;分离除去所述反应液中的水相,收集有机相,然后将所述有机相于70~75℃减压蒸馏除去甲苯得到2-乙醇酸甲酯-5-三氟甲基-1,3,4-噻二唑(化合物b1)230g,收率为95.0%,hplc检测含量为99.1%。
[0117]
(2)称取步骤(1)中全批产物2-乙醇酸甲酯-5-三氟甲基-1,3,4-噻二唑230g(化合物b1)、4-氟-n-异丙基苯胺146g(1.0eq)及甲苯(500ml)一次性加入2000ml三口瓶中,搅拌混合得到混合溶液;向所述混合溶液中加入20wt%氢氧化钠水溶液200ml,升温,在50℃的条件下,搅拌反应2h得到反应液;分离除去所述反应液中的水相,收集有机相,然后将所述有机相于70~75℃减压蒸馏除去甲苯收集固体,将固体烘干得到氟噻草胺330g,收率95.6%。hplc检测含量为99.2%。
[0118]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。