一种具有janus激酶抑制活性的化合物、包括该化合物的组合物及其应用
技术领域
1.本发明涉及医药领域,具体涉及一种具有janus激酶抑制活性的化合物、包括该化合物的组合物及其应用。
背景技术:
2.janus激酶抑制剂,也称为jak抑制剂或jakinibs
1.,是一种药物,通过抑制一种或多种janus激酶家族酶(jak1、jak2、jak3、tyk2)的活性来发挥作用,从而干扰/阻断jak-stat信号通路。jak-stat信号通路是近年来发现的一条由细胞因子刺激的信号转导通路,参与细胞的增殖、分化、凋亡以及免疫调节等许多重要的生物学过程。janus激酶(jak)抑制剂已被开发作为治疗以下疾病的药物,如血液系统疾病、肿瘤、类风湿性关节炎及白癜风、银屑病等炎症性疾病的治疗应用
[1][2]
,例如类风湿性关节炎
[3]
。人们对它们用于各种皮肤状况的用途很感兴趣
[4]
。
[0003]
jak-stat信号通路是一条由细胞因子刺激的信号转导通路,参与细胞的增殖、分化、凋亡以及免疫调节等许多重要的生物学过程。与其它信号通路相比,这条信号通路的传递过程相对简单,它主要由三个成分组成,即酪氨酸激酶相关受体、酪氨酸激酶jak和转录因子stat(信号转导和转录激活因子)。
[0004]
janus相关激酶介导了许多对造血和免疫功能很重要的细胞因子和生长因子的信号传导。jak信号传导涉及向细胞因子受体募集stat、激活和随后将stat定位到细胞核,从而调节基因表达。
[0005]
janus激酶抑制剂可以分为几个重叠的类别:免疫调节剂,dmard(疾病修饰抗风湿药),酪氨酸激酶抑制剂的一个子类。它们通过抑制细胞因子活性来改变免疫系统。细胞因子在控制细胞生长和免疫反应中起关键作用。许多细胞因子通过结合并激活i型和ii型细胞因子受体来发挥作用。这些受体又依赖于janus激酶家族进行信号转导。因此,抑制这些janus激酶活性的药物会阻断细胞因子信号传导
1.,比如说janus激酶磷酸化活化的细胞因子受体,这些磷酸化受体反过来募集调节基因转录的stat转录因子。
[0006]
jak/stat通路,特别是jak家族的所有四个成员,被认为在哮喘反应、慢性阻塞性肺病、支气管炎和其他相关的下呼吸道炎性疾病的发病机制中发挥作用
[5]
。jak/stat通路,尤其是jak3,也在免疫系统癌症中发挥作用,且抑制jak激酶对患有皮肤免疫疾病具有治疗益处。2020年fda批准一项新冠肺炎治疗药紧急使用批准(emergency use authorization,eua)——礼来jak抑制剂baricitinib联合瑞德西韦治疗需吸氧或通气住院治疗的新冠患者。
[0007]
ruxolitinib乳膏剂是incyte公司janus激酶抑制剂。2020年02月21日,incyte公司宣布,评估ruxolitinib乳膏剂治疗青少年和成人(年龄≥12岁)特应性皮炎(ad)患者iii期true-ad临床试验项目的第二个随机、赋形剂对照、关键性iii期true-ad1研究(nct03745638)已经达到了主要终点。特应性皮炎(ad)是一种以皮肤炎症和皮肤屏障缺陷
为特征的慢性皮肤病,影响全球多达10%的成年人和多达20%的儿童。
[0008]
美国哥伦比亚大学的研究人员,近日研发出一种新药ruxolitinib,或可令脱发秃头者重新长出头发。研究员指,秃头其中一个原因是免疫细胞破坏头发毛囊,而该种名为“ruxolitinib”的药物,或有助解决此问题。研究员找来3名秃头患者进行测试,效果良佳。3名测试者在4至5个月后,均长出浓密头发。然而研究尚在改进阶段,稍后需进行更多测试,以验证药物的安全。
[0009]
白癜风是一种比较常见的后天色素性皮肤病,表现为局限性或泛发性皮肤黏膜色素完全脱失。由于皮肤的黑素细胞功能消失引起,当产生黑色素的细胞死亡或停止运作时,就会发生白癜风,导致在皮肤色素沉着斑块损失。非节段性白癜风涉及色素脱失全身皮肤斑块。全身各部位可发生,常见于指背、腕、前臂、颜面、颈项及生殖器周围等。女性外阴部亦可发生,青年妇女居多。色素沉着丧失也常见于以下部位往往会经历摩擦、撞击或其他创伤,例如手和手臂。分段式白癜风与出现在一侧的小块脱色皮肤有关身体在有限的范围内。
[0010]
据估计,白癜风影响全球0.5%至2%的人口。白癜风的主要驱动因素是对黑色素细胞的自身免疫攻击,黑色素细胞是赋予皮肤颜色的细胞。这种自身免疫攻击的起始事件是未知的,但驱动黑素细胞破坏的关键细胞类型是cd8
溶细胞t细胞(ctl)。驱动这些ctl激活/功能的主要细胞因子是ifn-γ,其受体信号通过jak1和jak2传导。预计阻断jak1或jak2功能的药物可有效逆转黑色素细胞的自身免疫性破坏,这已通过使用托法替尼(一种jak抑制剂)和鲁索替尼(一种jak1抑制剂的初步研究得到证实)。
[0011]
新英格兰医学杂志(nejm)艾伯维jak1选择性抑制剂upadacitinib在与百时美施贵宝年销量29.8亿美元的阿巴西普(abatacept)头对头难治性类风湿性关节炎iii期临床中,显示出更好的疗效。upadacitinib,也是一种潜在的治疗风湿性关节炎和溃疡性结肠炎的jak1抑制剂。目前正针对多项适应症开展不同阶段的临床试验。
[0012]
ruxolitinib磷酸盐目前被批准用于治疗中危或高危骨髓纤维化患者,包括原发性骨髓纤维化、真性红细胞增多症后骨髓纤维化和原发性血小板增多症后骨髓纤维化。ruxolitinib磷酸盐目前还在临床试验中用于治疗原发性血小板增多症、胰腺癌、前列腺癌、乳腺癌、白血病、非霍奇金淋巴瘤、多发性骨髓瘤和银屑病。
[0013]
janus激酶抑制剂运用广泛。不断需要新的或改进的抑制激酶如janus激酶的药剂,作为器官移植的免疫抑制剂;以及预防和治疗自身免疫疾病的药剂(例如,多发性硬化症、类风湿性关节炎、哮喘、i、ii型糖尿病、炎症性肠病、克罗恩病、自身免疫性甲状腺疾病、阿尔茨海默病);涉及过度炎症反应的疾病的药剂(例如湿疹);涉及过敏、癌症(例如前列腺、白血病、多发性骨髓瘤)和一些免疫反应(例如,皮疹或接触性皮炎或腹泻)的药剂等等。本发明拟提供一种新的janus激酶抑制剂。
技术实现要素:
[0014]
为了克服现有技术中的缺陷,本发明提供了一种具有janus激酶抑制活性的化合物(4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-吡咯烷-1-酰胺化合物或其衍生物)、包括该化合物的药物组合物及其应用。
[0015]
为实现上述目的,本发明采用如下技术方案:
[0016]
本发明的第一方面是提供一种具有janus激酶抑制活性的化合物,其为如式i所示
的化合物或其衍生物,该衍生物为其药学上可接受的盐、酯、前药、单晶或多晶型物、溶剂化物或水合物;其中:
[0017][0018]
r1、r2各自独立地为氢原子或氘原子;
[0019]
r3为三氟甲基或三氟乙基或二氟乙氰基或(1-甲基-1-氟基)乙基,;
[0020]
r4为甲基或乙基或氟原子;
[0021]
r5为氢原子或氟原子;
[0022]
*是手性碳。
[0023]
进一步地,如式i所示的化合物包括如下结构式的化合物中的一种或多种:
[0024][0025]
(3s,4r)-3-氟基甲基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(c);
[0026][0027]
(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2-氟基-2-甲基丙基)吡咯烷-1-酰胺(d);
[0028][0029]
(3s,4r)-3-丙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(e);
[0030][0031]
(3s,4r)-3-乙基-4-(3h-5-氟基咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(f);
[0032][0033]
(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(3,3,3-三氟丙基)吡咯烷-1-酰胺(g);
[0034][0035]
(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2-二氟
丙氰基)吡咯烷-1-酰胺(h);
[0036][0037]
(3s,4s)-3-氟基甲基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(c2);
[0038][0039]
(3s,4s)-3-乙基-4-(3h-5-氟基咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(f2);
[0040][0041]
(3s,4r)-3-丙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,-二氟乙基)吡咯烷-1-酰胺(i);
[0042][0043]
(3s,4r)-3-氟基甲基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(3,3,3-三氟丙基)吡咯烷-1-酰胺(a);
[0044][0045]
(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(1,1-二氘-2,2,2-三氟乙基)吡咯烷-1-酰胺(b);
[0046][0047]
(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(1,1,2,2-四氘-3,3,3-三氟丙基)吡咯烷-1-酰胺(g-4d);
[0048][0049]
(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(1,1,-二氘-2,2-二氟丙氰基)吡咯烷-1-酰胺(h-2d)。
[0050]
本发明的第二方面是提供上述化合物在制备预防和/或治疗自身免疫疾病、炎性疾病、过敏、癌症、皮肤病、骨髓纤维化的药剂中的应用。
[0051]
进一步地,上述自身免疫疾病是自身免疫性皮肤障碍、多发性硬化症、风湿性关节炎、幼年关节炎、i型糖尿病、狼疮、炎症性肠病、克罗恩病、重症肌无力、免疫球蛋白肾病、心肌炎或自身免疫性甲状腺疾病。
[0052]
进一步地,上述自身免疫性皮肤障碍为斑秃、白癜风、特应性皮炎或银屑病。
[0053]
进一步地,上述皮肤病为脱发、特应性皮炎、牛皮癣、皮肤过敏、皮肤刺激、皮疹、接触性皮炎或接触过敏。
[0054]
进一步地,上述癌症为前列腺癌、肾癌、肝癌、乳腺癌、肺癌、甲状腺癌、卡波西肉瘤、卡斯尔曼病或胰腺癌、淋巴瘤、白血病、非霍奇金淋巴瘤、多发性骨髓瘤、皮肤t细胞淋巴瘤或皮肤b细胞淋巴瘤。
[0055]
进一步地,上述炎性疾病为虹膜炎、葡萄膜炎、巩膜炎、结膜炎、心肌炎、呼吸道的炎性疾病或炎性肌病。
[0056]
进一步地,上述骨髓纤维化为原发性骨髓纤维化、真性红细胞增多症后骨髓纤维化、原发性血小板增多症后骨髓纤维化、原发性血小板增多症或其组合。
[0057]
进一步地,上述药剂为固体制剂、半固体制剂或液体制剂。
[0058]
进一步地,上述固体制剂为片剂、胶囊剂、颗粒剂和丸剂的一种;上述半固体制剂为凝胶剂或软膏剂,上述液体制剂为注射剂、合剂或溶液剂。
[0059]
进一步地,上述药物的给药途径为口服给药、向腹膜内给药或静脉注射给药。
[0060]
本发明的第三方面是提供一种药物组合物,其包含治疗有效量的上述化合物以及药学上可接受的载体。
[0061]
本发明采用以上技术方案,与现有技术相比,具有如下技术效果:
[0062]
本发明提供的4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-吡咯烷-1-酰胺化合物或其衍生物具有优异的janus激酶抑制活性和优异的药效学性能,可作为janus激酶抑制剂为治疗相关疾病提供新的有效的选择。
附图说明
[0063]
图1是本发明一实施例中制备(3s,4r)-3-氟基甲基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(c)的流程图;
[0064]
图2是本发明一实施例中制备(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,-氟基-2-甲基丙基)吡咯烷-1-酰胺(d)的流程图;
[0065]
图3是本发明一实施例中制备(3s,4r)-3-丙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(e)的流程图;
[0066]
图4是本发明一实施例中制备(3s,4r)-3-乙基-4-(3h-5-氟基咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(f)的流程图;
[0067]
图5是本发明一实施例中制备(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,-二氟丙氰基)吡咯烷-1-酰胺(h)的流程图。
具体实施方式
[0068]
本发明提供了一种具有janus激酶抑制活性的化合物(4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-吡咯烷-1-酰胺化合物或其衍生物)、包括该化合物的药物组合物及其应用。该具有janus激酶抑制活性的化合物,其为一种如式i所示的化合物或其衍生物,该衍生物为其药学上可接受的盐(例如乙酸盐、酒石酸盐、三氟乙酸盐或磷酸盐)、酯、前药、单晶或多晶型物、溶剂化物或水合物;其中:
[0069][0070]
r1、r2各自独立地为氢原子或氘原子;
[0071]
r3为三氟甲基或三氟乙基或二氟乙氰基;
[0072]
r4为甲基或乙基或氟原子;
[0073]
r5为氢原子或氟原子;
[0074]
*是手性碳。
[0075]
具体地,作为上述如式i所示的化合物的衍生物中的一种,该化合物的药学上可接受的盐是指本发明的如式i所示的化合物与酸形成的无毒的、适于药用的盐,包括无机酸盐和有机酸盐。其中,适于形成无机酸盐的酸包括(但不限于)盐酸、氢溴酸、氢氟酸、硫酸、硝酸和磷酸等,而适于形成有机酸盐的酸包括(但不限于)甲酸、乙酸、丙酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、乳酸、苹果酸、柠檬酸、苦杏仁酸、葡糖酸、酒石酸、苦味酸、甲磺酸、亚甲基二磺酸、苯磺酸、对甲苯磺酸、天冬氨酸和谷氨酸等。
[0076]
具体地,作为上述如式i所示的化合物的衍生物中的一种,该化合物的前药是指通过各种机制(例如生物代谢、化学处理)在体内转化成本发明的如式i所示的化合物的物质。各种前药形式的实例可以参见下列文献:1)h.bundgaard,前药的设计和应用[m],1991,pp113-191;2)t.higuchi,w.stella,pro-drugs as novel delivery systems[m],a.c.s.symposium series,vol.14;和3)edward b.roche,bioreversible carriers in drug design[m],american pharmaceutical association and pergamon press,1987。
[0077]
具体地,作为上述如式i所示的化合物的衍生物中的一种,该化合物的溶剂化物是指本发明的如式i所示的化合物与有机溶剂因物理作用而形成的缔合物,包括(但不限于)乙醇化物和甲醇化物等。在某些特定情况下,当溶剂分子掺入结晶固体的晶格当中时,该溶剂化物就能够被分离纯化。
[0078]
具体地,作为上述如式i所示的化合物的衍生物中的一种,该化合物的水合物是指本发明的如式i所示的化合物与水因物理作用而形成的缔合物。
[0079]
溶剂化物和水合物的制备方法为本领域所熟知,典型的方法如下所述:在高于环境温度的条件下,将化合物溶解于所需量的溶剂(有机溶剂、水或二者的混合物)中,以足以形成结晶的速度冷却溶液,然后通过标准方法分离结晶,最后通过分析技术(例如红外光谱、热分析)来证实溶剂化物或水合物的结晶中溶剂或水的存在。
[0080]
介于上述化合物具有优异的janus激酶抑制活性,可用于预防和/或治疗自身免疫疾病、炎性疾病、过敏、癌症、皮肤病、骨髓纤维化的药剂。
[0081]
在本发明一优选的实施例式中,上述自身免疫疾病是自身免疫性皮肤障碍、多发性硬化症、风湿性关节炎、幼年关节炎、i型糖尿病、狼疮、炎症性肠病、克罗恩病、重症肌无力、免疫球蛋白肾病、心肌炎或自身免疫性甲状腺疾病。
[0082]
在本发明一优选的实施方式中,上述自身免疫性皮肤障碍为斑秃、白癜风、特应性皮炎或银屑病。
[0083]
在本发明一优选的实施方式中,上述皮肤病为脱发、特应性皮炎、牛皮癣、皮肤过敏、皮肤刺激、皮疹、接触性皮炎或接触过敏。
[0084]
在本发明一优选的实施方式中,上述癌症为前列腺癌、肾癌、肝癌、乳腺癌、肺癌、甲状腺癌、卡波西肉瘤、卡斯尔曼病或胰腺癌、淋巴瘤、白血病、非霍奇金淋巴瘤、多发性骨髓瘤、皮肤t细胞淋巴瘤或皮肤b细胞淋巴瘤。
[0085]
在本发明一优选的实施方式中,上述炎性疾病为虹膜炎、葡萄膜炎、巩膜炎、结膜炎、心肌炎、呼吸道的炎性疾病或炎性肌病。
[0086]
在本发明一优选的实施方式中,上述骨髓纤维化为原发性骨髓纤维化、真性红细胞增多症后骨髓纤维化、原发性血小板增多症后骨髓纤维化、原发性血小板增多症或其组合。
[0087]
在本发明一优选的实施方式中,所述药剂用于调节白癜风的一种或多种体征或症状。举例来说(但并非作为限制),白癜风的体征和症状包括(但不限于)皮肤(例如,面部、颈部、手、臂、脚、膝盖、生殖器和嘴唇上的皮肤)上的乳白色斑块产生(色素脱失),毛发(例如,头皮上的毛发、睫毛、眉毛或胡须)的过早白化或灰化,衬在口腔内部的组织的颜色损失,以及视网膜内层的颜色的损失或改变。因此,本发明公开提供治疗罹患白癜风的个体的方法。罹患白癜风的受试者可以通过例如本领域中已知的任何诊断或预后测定法来鉴别。
[0088]
在本发明一优选的实施方式中,上述药剂可以单独给药,或者与其他药学上可接受的化合物联合给药。
[0089]
本发明所述药剂可以是以任何可接受的剂型进行局部给药。在本发明一优选的实施方式中,上述药剂为固体制剂、半固体制剂或液体制剂。在本发明一优选的实施方式中,该药剂是以下形式施用:插入物、泵送系统、泡沫、凝胶、乳膏、洗剂、溶液、悬浮液、乳液、油膏、粉剂、晶体形式、脂质体、喷剂、糊剂、药膏、缓释纳米颗粒、二甲基亚砜(dmso)-基溶液悬
浮液、缓释微粒。
[0090]
在本发明一优选的实施方式中,上述固体制剂为片剂、胶囊剂、颗粒剂和丸剂的一种;上述半固体制剂为凝胶剂或软膏剂,上述液体制剂为注射剂、合剂或溶液剂。
[0091]
在本发明一优选的实施方式中,上述药剂可选任意给药途径给药,其包括经口服给药、静脉内或动脉内注射给药、脂肪内或脂质体给药、喷雾吸入给药、肠胃外或腹膜内给药、经直肠给药、皮下给药、脂肪内或关节内给药、经鼻给药、经由局部递送(例如导管或斯坦特氏印模)、含服给药、阴道给药或通过植入性药盒给药。优选的给药方式为口服给药、向腹膜内给药或静脉注射给药。
[0092]
在本发明中,术语“氘取代”或“氘代”表示化合物或基团中的一个或多个氢(h)被氘取代,氘取代可以是一取代、二取代、多取代或全取代。术语“氢”表示单个h原子。术语“氘”表示单个氘原子。例如,单氘代甲基可以表示为-cdh2,二氘代甲基可以表示为-cd2h,三氘代甲基可以表示为-cd3。
[0093]
下面通过具体实施例和附图对本发明进行详细和具体的介绍,以使更好的理解本发明,但是下述实施例并不限制本发明范围。
[0094]
实施例中方法如无特殊说明的采用常规方法,使用的试剂如无特殊说明的使用常规市售试剂或按常规方法配制的试剂。
[0095]
在以下实施例中,除非另有说明,否则温度以摄氏度(℃)表示;操作在室温或环境温度下进行,“rt”或“rt”(通常范围为18-25℃);溶剂的蒸发使用减压(通常为4.5-30毫米汞柱)下的旋转蒸发器进行,浴温高达60℃;反应过程通常采用薄层色谱法(tlc)跟踪;熔点未修正;产品显示出令人满意的1h-nmr和/或微量分析数据;使用以下常规缩写:l(升)、ml(毫升)、mmol(毫摩尔)、g(克)、mg(毫克)、min(分钟)和h(小时)。
[0096]
实施例1
[0097]
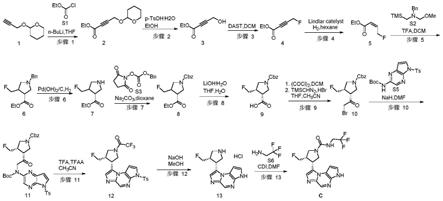
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-氟基甲基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(c),其制备流程如下(其中化合物c的合成路线图如图1):
[0098]
步骤1:
[0099][0100]
氮气保护下,化合物1(10.0g,71.3mmol,1.0eq)的无水thf(200ml)溶液用干冰-丙酮浴冷却至-68℃。向上述溶液中慢慢滴加n-buli(29.1ml,2.5m in hexane(己烷),72.8mmol,1.05eq),45分钟滴加完毕。随后,在-65℃搅拌1小时。向上述反应液滴加化合物s1(11.6g,107.0mmol,1.5eq)。滴毕,-65℃搅拌0.5小时。tlc检测显示反应完毕。
[0101]
反应液经氯化铵水溶液(100ml)淬灭。分出有机相。水相用乙酸乙酯(30ml
×
2)萃取2次。合并有机相,食盐水(100ml
×
2)洗涤。无水硫酸钠干燥,过滤,浓缩,得到14.5g黄色油状物。收率:95.8%。分析数据如下:
[0102]
hplc纯度:86.1%(220nm);lcms(esi ):m/z 235.2(m na);1h-nmr(300mhz,cdcl3)
4.83~4.82(m,1h),4.40~4.39(m,2h),4.26(q,j=7.2hz,2h),3.87~3.79(m,1h),3.58~3.53(1h),1.78~1.66(m,3h),1.60~1.53(m,3h),1.31(t,j=7.2hz,3h).
[0103]
步骤2:
[0104][0105]
向化合物2(14.5g,68.3mmol,1.0eq)的乙醇(45ml)溶液中加入p-tsoh
·
h2o(390mg,2.0mmol,3%eq)。升温回流,反应3.5小时。冷至室温后,搅拌1.5小时。hplc显示反应完毕。向反应体系中加入10ml nahco3水溶液和100毫升水。反应混合物用dcm萃取(30ml
×
3)。合并有机相,食盐水洗涤(40ml
×
2)。无水硫酸钠干燥,过滤,浓缩,得17.5克粗品(3750-019-p1)。
[0106]
该粗品(3750-019-p1)经硅胶柱层析纯化(石油醚/乙酸乙酯=10/1~5/1)。得6.8g(3750-019-p2)黄色油状物。收率:77.7%。分析数据如下:
[0107]
hplc纯度:98.3%(at 220nm);lcms(esi ):m/z 129.1(m h);1h-nmr(300mhz,cdcl3)4.39(s,2h),4.25(q,j=7.2hz,2h),1.28(t,j=7.2hz,3h).
[0108]
步骤3:
[0109][0110]
氮气保护下,dast(5.3g,33.2mmol,0.85eq)的dcm溶液(25ml)用干冰丙酮浴冷却至-65℃。化合物3(5.0g,39.0mmol,1.0eq)滴加至上述溶液。滴加时间为25分钟。滴毕,-65℃搅拌2小时。升至室温,并搅拌过夜。hplc显示剩余2.5%化合物3。反应体系用水洗涤(20ml
×
3),无水硫酸钠干燥,过滤,浓缩,得4.8克粗品(3750-035-p1)。
[0111]
该粗品(3750-035-p1)经减压蒸馏提纯,得2.0克纯品(3750-035-p2),无色油状物。收率:39.4%。分析数据如下:
[0112]
hplc纯度:97.7%(220nm);1h-nmr(300mhz,cdcl3)5.16(s,1h),5.00(s,1h),4.25(q,j=7.2hz,2h),1.26(t,j=7.2hz,3h).
[0113]
步骤4:
[0114][0115]
向化合物4(2.0g,15.4mmol,1.0eq)的正己烷溶液(20ml)中加入lindlar催化剂(318mg,1.54mmol,10%eq)。氢气置换3次后,室温搅拌6小时。hplc显示反应完毕。经硅藻土过滤后,由于产品易挥发,所以滤液未经浓缩,直接用于下一步反应。
[0116]
步骤5:
044-p2),无色油状物。收率:63.3%。分析数据如下:
[0129]
hplc纯度:95.7%(210nm);lcms(esi ):m/z 332.1(m na);1h-nmr(300mhz,cdcl3)7.37~7.30(m,5h),5.20~5.10(m,2h),4.63~4.36(m,2h),4.17(q,j=7.2hz,2h),3.78~3.59(m,3h),3.56~3.51(m,1h),3.22~3.17(m,1h),2.91~2.80(m,1h),1.27(t,j=7.2hz,3h).
[0130]
步骤8:
[0131][0132]
化合物8(1.2g,3.9mmol,1.0eq)的thf(12ml)和水(12ml)的混合溶液冷却至零度,加入lioh
·
h2o(163mg,3.9mmol,1.0eq),并在零度搅拌1.5小时。
[0133]
hplc显示反应完全。向体系中加入水(12ml),并用mtbe洗涤水相(12ml
×
2)。分去有机相,用2m hcl将水相ph调至1,并用乙酸乙酯萃取(10ml
×
3)。合并有机相,食盐水洗涤(10ml)。无水硫酸钠干燥,过滤,浓缩,得1.09g粗品(3750-052-p1)。hplc显示cis/trans~9/2。
[0134]
该粗品(3750-052-p1)经硅胶柱层析纯化(dcm/meoh=1/0~100/1),得1.0g产品(3672-052-p2)。该产品进一步经超临界流体色谱纯化,得410mg产品(构型确认见专利wo2019016745a1)。该产品经制备hplc进一步纯化,得218mg产品(3750-052-p3-peak 1)。
[0135]
hplc纯度:96.1%(210nm);lcms(esi ):m/z 304.1(m na);1h-nmr(300mhz,cdcl3)7.38~7.30(m,5h),5.20~5.10(m,2h),4.70~4.43(m,2h),3.80~3.62(m,3h),3.57~3.54(m,1h),3.27~3.21(m,1h),2.93~2.85(m,1h).
[0136]
步骤9:
[0137][0138]
氮气保护下,零度,向化合物9(174mg,0.62mmol,1.0eq)的dcm(2ml)溶液中滴加(cocl)2(157mg,1.24mmol,2.0eq)的dcm(1ml)溶液。10分钟滴毕。再搅拌10分钟,加入催化量的dmf。升至室温后搅拌过夜。
[0139]
将反应液浓缩至干后,加入1.5ml的ch3cn和1.5ml的thf。并冷却至-18℃。滴加tmschn2(1.24ml,2.48mmol,2m in hexane,4.0eq),10内滴加完毕。然后在不超过-10℃下搅拌3小时。
[0140]
向体系中滴加hbr水溶液(0.7ml,6.2mmol,40%wt,10.0eq),30分钟内滴加完毕。继续搅拌30分钟,hplc显示反应完毕。向反应中滴加水(10ml)淬灭反应。然后用乙酸乙酯萃取(5ml
×
3)。合并有机相,并有食盐水洗涤(5ml
×
3)。无水硫酸钠干燥,过滤,浓缩,得155mg产品(3750-061-p1)。收率:70.0%。
[0141]
hplc纯度:88.4%(210nm);lcms(esi ):m/z 380.0,382.1(m na);1h-nmr(300mhz,cdcl3)7.36~7.32(m,5h),5.19~5.09(m,2h),4.55~4.28(m,2h),4.05~3.89(m,2h),3.79~3.61(m,4h),3.45~3.41(m,1h),3.10~2.98(m,1h).
[0142]
步骤10:
[0143][0144]
氮气保护下,60%nah(15mg,0.37mmol,1.0eq)的无水dmf(1ml)溶液冷却至-20℃。化合物s5(174mg,0.49mmol,1.2eq)的无水dmf(1ml)溶液滴加至上述反应体系中。15分钟滴加完毕。然后继续搅拌2小时。将此反应液在-20℃下分批加至化合物10(134mg,0.37mmol,1.0eq)的无水dmf(1ml)溶液中。加入1.5ml时,hplc显示反应完全。nh4cl水溶液(2ml)滴加至反应中淬灭。用乙酸乙酯萃取(3ml
×
3),合并有机相,并用食盐水洗涤(3ml
×
2)。无水硫酸钠干燥,过滤,浓缩,得275mg粗品。
[0145]
该粗品(3750-065-p1)经制备硅胶色谱提纯,得100mg白色固体。收率:34.6%。分析数据如下:
[0146]
hplc纯度:98.4%(254nm);lcms(esi ):m/z 666.0(m h);1h-nmr(300mhz,cdcl3)8.88(s,1h),8.01(d,j=8.4hz,2h),7.89~7.86(m,1h),7.36~7.29(m,7h),6.97~6.64(m,1h),5.13~5.09(m,2h),4.76~4.66(m,2h),4.58~4.36(m,2h),3.95~3.79(m,1h),3.70~3.43(m,4h),3.01~2.88(m,1h),2.38(s,3h),1.51(s,9h).
[0147]
步骤11:
[0148][0149]
化合物11(116mg,0.17mmol,1.0eq)的ch3cn溶液(2.5ml)冷却至0℃。并加入0.12ml的tfa和0.62ml的tfaa。然后升温至55℃并搅拌24h。hplc剩余7%的中间体12-int。补加0.2ml的tfaa并继续搅拌15h。hplc反应完毕。将反应浓缩至干,并加入5ml的乙酸乙酯。加入nahco3水溶液调节ph至9。分出有机相,水相用乙酸乙酯萃取(3ml
×
2)。合并有机相,并用食盐水洗涤(5ml
×
2)。无水硫酸钠干燥,过滤,浓缩,得135mg粗品(3750-068-p1)。
[0150]
该粗品(3750-068-p2)经制备硅胶色谱提纯,得78mg黄色固体产品。收率:87.9%。
[0151]
hplc纯度:91.8%(220nm);lcms(esi ):m/z 509.9(m h);1h-nmr(300mhz,cdcl3)
8.84(s,1h),8.12(d,j=8.4hz,2h),7.79(d,j=3.9hz,1h),7.65(d,j=1.5hz,1h),7.35(d,j=8.4hz,2h),6.84(d,j=3.6hz,1h),4.44(d,j=5.7hz,2h),4.24~4.19(m,1h),4.17~4.01(m,5h),2.40(s,3h).
[0152]
步骤12:
[0153][0154]
化合物12(6.8mg,0.013mmol,1.0eq)的meoh(0.5ml)溶液冷却至0℃。向其中加入0.25ml的1m naoh水溶液。然后室温搅拌2小时。tlc显示反应完毕。加入2m hcl调节ph至2。将反应浓缩干,并加入2ml的meoh,过滤除去不溶物。滤液再次浓缩,得16mg粗品,该粗品未经纯化直接用于下一步。
[0155]1h-nmr(300mhz,cd3od)8.94(s,1h),8.44(s,1h),7.90(d,j=3.3hz,1h),7.35(d,j=3.6hz,1h),4.34(s,2h),4.04~3.91(m,3h),3.71~3.53(m,3h).
[0156]
步骤13:
[0157][0158]
向cdi(37.3mg,0.23mmol,1.5eq)的无水dmf(1ml)溶液中加入et3n(31mg,0.31mmol,2.0eq)的无水dmf(0.7ml)溶液。并冷却至0℃。向上诉体系中滴加化合物s6(18.2mg,0.18mmol,1.2eq)的无水dmf(0.6ml)溶液。滴毕,升至室温,并搅拌1.5小时。将此反应液在室温下分批加至化合物13(120mg,0.15mmol,1.0eq)的无水dmf(2ml)溶液中。加入2.0ml时,lcms显示反应完毕。加入水(15ml)淬灭反应。并用dcm/meoh=5/1(5ml
×
4)萃取。合并有机相,并用食盐水洗涤(5ml
×
2)。无水硫酸钠干燥,过滤,浓缩,得90粗品(3750-074-p1)。
[0159]
该粗品(3750-074-p1)经制备硅胶色谱纯化,得14mg类白色固体。
[0160]
hplc纯度:99.6%(254nm);lcms(esi ):m/z 384.8(m h);1h-nmr(300mhz,cd3od)8.59(s,1h),7.65(s,1h),7.45(d,j=2.4hz,1h),7.03(d,j=2.7hz,1h),4.62~4.60(m,1h),4.29~4.21(m,1h),4.16~3.87(m,6h),3.67~3.63(m,1h),3.25~3.18(m,1h).
[0161]
实施例2
[0162]
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2-氟基-2-甲基丙基)吡咯烷-1-酰胺(d),其制备流
程如下(如图2):
[0163]
步骤1:
[0164][0165]
在氮气保护下,将nah(0.345g,8.61mmol,1.0eq)缓慢加入至无水dmf(10ml)中,冰盐浴降温至0℃。向上述体系中缓慢滴加化合物a(3.35g,8.61mmol,1.0eq)的dmf溶液10ml,控制加料温度始终不超过0℃,加完继续保温反应30分钟。然后体系继续滴加化合物d-1(3.05g,8.61mmol,1.0eq)的dmf溶液10ml,加完保温0℃继续搅拌1.5小时。tlc显示原料d-1消耗完全。用0.2l冰水淬灭反应,并用乙酸乙酯(50ml
×
3)萃取,食盐水(40ml
×
2)洗涤合并的有机层,无水硫酸钠干燥,过滤、浓缩得到6.8克粗品。经过硅胶柱纯化后,得到3.9g化合物d-2,半油状固体。并得到1.1g副产物,为白色固体。产率:68.5%。lcms(esi ):m/z 662.0(m h)。
[0166]
步骤2:
[0167][0168]
在氮气保护下,将化合物d-2(3.6g,5.4mmol,1.0eq)溶于二氯甲烷(40ml)中并降温至10℃左右。缓慢向体系滴加三氟乙酸5ml。加完体系自然升温至室温并继续反应2.5小时。直接浓缩反应,得到2.8g化合物d-3,直接进入用于下一步,无需纯化。产率:82%。lcms(esi ):m/z 562.2(m h)。
[0169]
步骤3:
[0170][0171]
在氮气保护下,将化合物d-3(2.0g,3.56mmol,1.0eq)溶于无水乙腈(20ml)中,依次加入三氟乙酸(0.5ml)和三氟乙酸酐(5ml)。反应体系加热至55℃并持续保温反应48小时。tlc显示原料反应完全。减压除去大部分溶剂,剩余物用100ml碳酸氢钠溶液中和淬灭,再用乙酸乙酯(50ml
×
3)萃取,饱和食盐水(40ml
×
2)洗涤合并的有机层,无水硫酸钠干燥,过滤、浓缩得2.3g棕黄色油状的粗品化合物d-4,无需进一步纯化直接用于下一步。lcms(esi ):m/z 506.1(m h);
[0172]
注:苯甲醇作为主要杂质残留在粗产品中。
[0173]
步骤4:
[0174][0175]
在氮气保护下,将化合物d-3(2.0g,3.96mmol,1.0eq)溶于乙腈中(20ml)并降温至10℃左右。缓慢向体系滴加naoh溶液(10mmol,2.5eq,0.4g,溶于10ml水)。加完后自然升温至室温搅拌2小时。lc-ms跟踪,原料10消耗完全,低温浓缩除去大部分溶剂。然后用6m hcl酸化至ph=1-2。用乙酸乙酯(15ml
×
2)洗涤水溶液,收集水层并浓缩除水。然后将所得残留物溶于20ml meoh中并过滤除去无机盐。浓缩滤液,得到1.8g粗品int-11,直接用于下一步,无需纯化。产率:150%。lcms(esi ):m/z 256.1(m h)。
[0176]
步骤5:
[0177]
[0178]
在氮气保护下,将cdi(305mg,1.97mmol,1.0eq)溶于dmf(10ml)中,冰水浴降温至10℃。然后向体系缓慢滴加化合物a(200mg,1.97mmol,1.0eq)的dmf溶液1ml,加完继续反应1.5小时。同时在n2保护下,将化合物int-11(800mg,0.4eq)溶于dmf(10ml)中,并加入tea(1ml,7.2mmol,3eq)进行游离底物。将新配置的含有化合物int-11的溶液缓慢逐滴加入到含有化合物a的溶液中,加完继续反应3小时。tlc跟踪,原料11消耗完全。用100ml冰水淬灭反应,并用ea(50ml
×
3)萃取。用食盐水(40ml
×
2)洗涤合并的有机层,并用无水na2so4干燥。过滤浓缩后,所得残留物通过硅胶柱纯化,得到220mg目标产品d。
[0179]
lc-ms(esi ):m/z 373.1(m h);1h-nmr(300mhz,cdcl3):δ8.73(s,1h),7.57(s,1h),7.30(s,1h),6.77(s,1h),4.70(m,2h),4.27(m,1h),3.82-4.03(m,2h),3.71-3.77(t,j=7.2hz,1h),3.56(d,j=6.0hz,1h),3.46(d,j=6.0hz,1h),3.39(t,j=6.9hz,1h),2.65(m,1h),1.44(s,3h),1.37(s,3h),0.85-1.01(m,2h),0.76(t,j=7.2hz,3h).
[0180]
实施例3
[0181]
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-丙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(e),其制备流程如下(如图3):
[0182]
步骤1:
[0183][0184]
氮气保护下,化合物es1(18.0g,264.2mmol,1.0q)的无水thf(500ml)溶液经干冰丙酮浴冷却至-65℃。向上述反应中滴加n-buli(116ml,2.5m in hexane,290.7mmol,1.1eq)溶液。50min滴加完毕。然后保温反应1小时。将化合物乙基氯甲酸酯(ethylcarbonochloridate,37.3g,343.5mmol,1.3eq)在-65℃下滴加至上述反应中,30min滴加完毕。然后继续搅拌0.5小时。tlc显示反应完全。
[0185]
在0℃加入10%nh4cl水溶液(250ml)淬灭反应。分离有机相,并用mtbe(200ml
×
2)萃取水层。合并有机相,并用盐水(400ml
×
2)洗涤。无水na2so4干燥,过滤,浓缩,得到37.0g化合物e1,黄色油状物。收率:99.9%。分析数据如下:
[0186]
1h-nmr(300mhz,cdcl3)4.25-4.18(m,2h),2.34-2.29(m,2h),1.68-1.52(m,2h),1.28(t,j=7.2hz,3h),0.97(t,j=7.8hz,3h).
[0187]
步骤2:
[0188][0189]
向化合物e1(20.0g,142.8mmol,1.0eq)的己烷(400ml)溶液中加入lindlar催化剂(2.9g,14.3mmol,10%eq)。用h2置换三次,并在rt下搅拌3h。hplc显示反应完全。经过滤浓
缩,得到21.5克化合物e2,黄色油状物。收率:106.0%。分析数据如下:
[0190]
1h-nmr(300mhz,cdcl3)6.26-6.17(m,1h),5.77(d,j=11.4hz,1h),4.25-4.18(m,2h),2.61(dd,j=7.5hz,2.4hz,2h),1.49-1.41(m,2h),1.32-1.28(m,3h),0.95(t,j=7.5hz,3h).
[0191]
步骤3:
[0192][0193]
在n2保护下,将tfa(170mg,1.5mmol,1%eq)在-15℃下添加到化合物e2(21.2g,149.2mmol,1.0eq)和s2(35.4g,149.2mmol,1.0eq)的dcm(150ml)溶液中。反应在-10℃~-20℃下搅拌5小时,然后加热至室温过夜。hplc显示化合物s2已被消耗完。添加20%nahco3溶液(300ml)淬灭反应。分离有机相,用dcm(200ml
×
2)萃取水层。用食盐水(300ml
×
1)洗涤合并的有机层。无水na2so4干燥,过滤,浓缩,得38.1g粗品e3,黄色油状物。
[0194]
粗产物(3714-010-p1)通过硅胶柱(石油醚/乙酸乙酯=20/1~5/1)纯化。得25.7g产品,为黄色油。收率:62.6%。分析数据如下:
[0195]1h-nmr(300mhz,cdcl3)7.32-7.26(m,5h),4.18-4.13(m,2h),3.65(s,2h),3.14-3.06(m,3h),2.67-2.61(m,1h),2.52-2.48(m,1h),1.16-1.09(m,1h),1.32-1.22(m,5h),0.91-0.89(m,3h).
[0196]
步骤4:
[0197][0198]
向化合物e3(36.3g,131.9mmol,1.0eq)甲醇(218ml)溶液中添加pd(oh)2/c(7.3g,占化合物e3质量的20%)。用h2置换三次,并在室温下搅拌过夜。hplc分析显示化合物e3已消耗完。混合物通过硅藻土过滤,滤饼用甲醇(50ml
×
2)淋洗。将滤液浓缩,得到29g棕色油状粗品e4。
[0199]
粗产物经硅胶柱(dcm/meoh=50/1~10/1)纯化,得19.0g产品,为黄色油。收率:77.8%。
[0200]1h-nmr(300mhz,cdcl3)4.20-4.15(m,2h),3.42-3.38(m,1h),3.28-3.20(m,2h),3.08-3.03(m,1h),2.89-2.82(m,1h),2.49-2.43(m,1h),1.37-1.25(m,7h),0.91(t,j=6.6hz,3h).
[0201]
步骤5:
[0202][0203]
将e4(7.6g,41.1mmol,1.0eq)和6n hcl(84ml)的混合物加热至100℃搅拌16h。lcms分析表明化合物e4已被消耗完。将反应混合物在55℃下减压浓缩,得8.9g黄色固体粗品化合物e5。
[0204]1h-nmr(300mhz,cdcl3)9.56(brs,1h),9.28(brs,1h),3.63-3.31(m,3h),3.29-3.12(m,1h),2.92-2.79(m,1h),2.51-2.42(m,1h),1.36-1.29(m,4h),0.87(t,j=6.0hz,3h).
[0205]
步骤6:
[0206][0207]
向化合物e5(8.9g,56.6mmol,1.0eq)的二氧六环(135ml)溶液中加入na2co3(8.4g,79.3mmol,1.4eq)。将混合物冷却至0℃,并分批加入化合物s3(17.0g,68.0mmol,1.2eq)。然后,将混合物加热至室温并搅拌过夜。tlc显示化合物e5已被消耗完。
[0208]
向反应中加入水(150ml),并用乙酸乙酯萃取(150ml
×
2)。用饱和na2co3(100ml
×
2)洗涤合并的有机层。将水层用6n hcl酸化至ph 3~4,并用乙酸乙酯(200ml
×
2)萃取。用水(200ml)和盐水(200ml)洗涤合并的有机层。无水na2so4干燥,过滤,浓缩,得到14.2g粗品e6,为黄色油状物。
[0209]
粗产物经硅胶柱(dcm/meoh=50/1~30/1)纯化。得9.5g产品e6,为无色油。hplc分析显示顺式/反式为9/2。通过sfc进一步纯化产物,得3.5g产物e7。分析数据如下:
[0210]1h-nmr(300mhz,cdcl3)7.36-7.26(m,5h),5.13(dd,j=12.0hz,17.4hz,2h),3.75-3.53(m,3h),3.30-3.21(m,1h),3.05-3.01(m,1h),2.42-2.38(m,1h),1.36-1.29(m,4h),0.90(t,j=6.6hz,3h).
[0211]
步骤7:
[0212][0213]
氮气气氛下,向e7(3.28g,11.3mmol,1.0eq)的dcm溶液中,在0℃滴加2m(cocl)2(2.86g,22.6mmol,2.0eq)的dcm(11.3ml)溶液。然后,搅拌10分钟,并加入一滴dmf。将混合
物加热至室温并搅拌过夜。
[0214]
将混合物浓缩至干,用acn/thf(16.4ml/16.4ml)溶解残余物。所得溶液在氮气气氛下冷却至-10℃。将tmschn2(22.6ml,45.3mmol,4.0eq)在-5℃下逐滴加入到上述溶液中。并在-10~-5℃下搅拌3小时。然后在-5℃下滴加47%hbr(13.0ml,113mmol,10.0eq)。滴毕,温度上升至10℃,并在此温度下搅拌2小时。
[0215]
tlc显示反应完成后,将混合物冷却至0℃并用水(100ml)淬灭。用mtbe(100ml,50ml)萃取两次。用nahco3水溶液(70ml)和食盐水(70ml)洗涤合并的有机层。无水硫酸钠干燥,浓缩,得4.2g粗产物,将其通过硅胶柱(dcm/meoh 30/1)纯化,得3.4g黄色油状物不纯产物。收率:82.0%。
[0216]1h-nmr(300mhz,cdcl3)7.41-7.32(m,5h),5.18-5.07(m,2h),3.92-3.84(m,2h),3.74-3.52(m,5h),2.52-2.42(m,1h),1.36-1.22(m,4h),0.94-0.86(m,3h).
[0217]
步骤8:
[0218][0219]
氮气气氛下,在-5℃向60%nah(326mg,8.15mmol,1.0eq)的dmf(15ml)混合物中,滴加s4(3.16g,8.15mmol,1.0eq)dmf(15ml)溶液。滴毕,在-5℃下搅拌90分钟。然后滴加e8(3.0g,8.15mmol,1.0eq)的dmf(15ml)溶液。滴毕,将混合物在-5℃下搅拌60分钟。
[0220]
tlc显示反应完成后,将混合物淬灭至冰水(150ml)中。用乙酸乙酯(所采用的的体积分别为150ml,70ml,70ml)萃取3次。用食盐水(100ml
×
3)洗涤合并的有机层。无水硫酸钠干燥,过滤,浓缩,得7.1g粗品。用硅胶柱(己烷/乙酸乙酯9/1~3/1)纯化,得3.7g产品。收率:59.3%。分析数据如下:
[0221]1h-nmr(300mhz,cdcl3)8.89(s,1h),8.00(d,j=8.1hz,2h),7.88-7.84(m,1h),7.33-7.26(m,7h),6.55(dd,j=4.2hz,46.8hz,1h),5.12-5.07(m,2h),4.73(dd,j=18.0hz,31.2hz,2h),3.82-3.72(m,1h),3.63-3.53(m,2h),3.43-3.25(m,2h),1.52-1.41(m,1h),2.38(s,3h),1.59(s,9h),1.39-1.28(m,4h),0.93-0.84(m,3h).
[0222]
步骤9:
[0223][0224]
在10℃下,将tfa(3.5ml)逐滴加到e9(3.5g,5.18mmol,1.0eq)的混合物中。再加入tfaa(17.5ml)。将混合物加热至55℃并搅拌过夜。再加入tfaa(7.5ml)并搅拌6小时。浓缩,用乙酸乙酯(100ml)和水(100ml)稀释残余物,然后用nahco3水溶液碱化至ph 8-9。分离有机层,用乙酸乙酯(100ml,50ml)萃取水层。用nahco3水溶液(150ml)和食盐水(150ml)洗涤合并的有机层。无水硫酸钠干燥,过滤,浓缩,得3.7g粗产品,为黄色油。通过硅胶柱(己烷/乙酸乙酯~3/1)纯化,得到2.2g产品。收率:81.7%。
[0225]1h-nmr(300mhz,dmso-d6)8.84(s,1h),8.12(d,j=8.4hz,2h),7.79(d,j=3.6hz,1h),7.55(d,j=7.2hz,1h),7.34-7.26(m,2h),6.69-6.67(m,1h),4.44(d,j=5.4hz,1h),4.26-3.98(m,3h),3.97-3.95(m,1h),2.86-2.62(m,1h),2.40(s,3h),1.28-0.97(m,3h),0.92-0.81(m,1h),0.76-0.65(m,3h).
[0226]
步骤10:
[0227][0228]
向e10(1.7g,3.3mmol,1.0eq)的甲醇(34ml)溶液中滴加1n naoh(17ml,17mmol,5.0eq),温度控制为0-5℃。滴毕,搅拌1小时。tlc表明反应完成。用2n hcl将混合物酸化至ph=2,然后浓缩。残余物用甲醇溶解并过滤以除去无机盐。滤液浓缩至干,用甲醇溶解,然后过滤去除无机盐。滤液浓缩至干,得1.9g粗产品,无需进一步纯化,直接用于下一步。
[0229]
步骤11:
[0230][0231]
氮气气氛下,5℃,向cdi(797mg,4.9mmol,0.9eq)和et3n(993mg,9.8mmol,1.8eq)的dmf(30ml)溶液中滴加(2,2,2-三氟乙基)氨(2,2,2-trifluoroethan-1-amine,389mg,3.9mmol,0.7eq)的dmf(2ml)溶液。滴毕,将混合物加热至室温并搅拌30分钟,然后在室温下滴加e11(粗品,1.9g,5.5mmol,1.0eq)的dmf(30ml)溶液。滴毕,将混合物搅拌约3小时,直到tlc分析显示大部分e11被消耗。
[0232]
将混合物倒入冰水(200ml)中,并用乙酸乙酯(200ml
×
3)萃取。用盐水(200ml
×
3)洗涤合并的有机层。无水硫酸钠燥,过滤,浓缩,得1.6g粗产品,为黄色油。通过硅胶柱(dcm/meoh~98/2)纯化,得到225mg产品e。
[0233]
hplc纯度:99.7%(254nm);lcms(esi ):m/z 395.2(m h);1h-nmr(300mhz,cdcl3)9.98(brs,1h),8.72(s,1h),7.56(s,1h),7.31(s,1h),6.76(s,1h),4.75-4.73(m,1h),4.29-4.21(m,1h),4.02-3.89(m,4h),3.78-3.72(m,1h),3.42-3.37(m,1h),2.81-2.74(m,1h),1.32-1.21(m,2h),1.17-1.11(m,1h),0.97-0.82(m,1h),0.71(t,j=6.3hz,3h).
[0234]
实施例4
[0235]
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-乙基-4-(3h-5-氟基咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(f)和(3s,4s)-3-乙基-4-(3h-5-氟基咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(f2),其制备流程如下(其中化合物f的合成路线图如图4):
[0236]
步骤1:
[0237][0238]
在氮气保护下,将化合物f-1(100mg,0.505mmol,1.0eq)溶于乙腈/醋酸(1ml/1ml)中,加入selectfluor试剂(179mg,0.505mmol,1.0eq),反应室温搅拌2小时。补加0.3eq的selectfluor试剂,反应继续搅拌5小时。tlc跟踪,绝大部分原料f-1基本消耗。用15ml h2o淬灭反应,乙酸乙酯(15ml
×
2)萃取,食盐水(20ml
×
2)洗涤合并的有机层,并用无水na2so4干燥。过滤和浓缩后,残留物通过硅胶柱纯化,得到60mg的化合物f-2。产率:55%。
[0239]
lcms(esi ):m/z 216.0(m h);1h-nmr(300mhz,cdcl3):δ8.53(brs,1h),8.50(s,1h),7.42(t,j=2.4hz,1h).
[0240]
步骤2:
[0241][0242]
化合物f-2(1.8g,8.33mmol,1.0q)溶于无水dmf(18ml)溶液中,降温至0℃。分批加入nah(500mg,12.5mmol,1.5eq,60%),反应体系保温0℃反应1小时。tscl(1.9g,10.0mmol,1.2eq)的dmf(9ml)溶液缓慢滴加到上述溶液中,加完后体系自然升温并继续反应1.5h。tlc跟踪,原料f-2消耗完全。用100ml冰水淬灭反应,体系析出大量固体。过滤,用10ml水洗涤滤饼。收集滤饼并溶于30ml乙酸乙酯中,并用无水na2so4干燥。过滤并浓缩后,得到3.1g化合物f-3,无需纯化直接用于下一步。产率:100.5%。
[0243]
lcms(esi ):m/z 370.0(m h);1h-nmr(300mhz,cdcl3):δ8.47(s,1h),8.00(d,j=8.4hz,2h),7.82(d,j=2.1hz,1h),7.32(d,j=8.1hz,2h),2.40(s,3h).
[0244]
步骤3:
[0245][0246]
在氮气保护下,化合物f-3(2.7g,7.3mmol,1eq)溶解于1,4-二恶烷(54ml)中,依次加入bocnh2(1.3g,10.9mmol,1.5eq)、k2co3(3.0g,21.9mmol,3eq)和pd(oac)2(163.5mg,0.73mmol,0.1eq)。体系室温下搅拌反应1h。tlc跟踪,原料f-3消耗完全。用100ml水淬灭反应,乙酸乙酯(50ml
×
3)萃取,食盐水(30ml
×
3)洗涤合并的有机层,并用无水na2so4干燥。过滤浓缩后得到粗产物4.0g,柱层析(pe/ea=20/1~5/1)纯化得到2.7g化合物f-4。产率:91%。
[0247]
lcms(esi ):m/z 407.1(m h);1h-nmr(300mhz,cdcl3):δ9.16(s,1h),7.99(d,j=8.4hz,2h),7.71(d,j=2.1hz,1h),7.41(m,1h),7.24(m,1h),2.28(s,3h),1.55(s,9h).
[0248]
步骤4:
[0249][0250]
在氮气保护下,nah(112mg,2.8mmol,60%eq)溶于无水dmf(11ml)中并降温至-15℃。向体系缓慢滴加化合物f-4(1.14g,2.8mmol,1eq)的dmf(11ml)溶液,加完-15℃继续反应1h。然后缓慢向体系滴加化合物fs1(1.0g,2.8mmol,1eq)的dmf溶液6ml,控制滴加时温度
在-15℃左右,滴加完毕后,保温搅拌反应1小时,然后自然升温至室温并搅拌40分钟。tlc跟踪,原料f-4消耗完全。用1m的nh4cl溶液(64ml)淬灭反应,乙酸乙酯(50ml
×
3)萃取,食盐水(20ml
×
2)洗涤合并的有机层,并用无水na2so4干燥。过滤浓缩后得到粗产物2.1g,柱层析(pe/ea=10/1~1/1)纯化得到1.1g化合物5。产率:56%。
[0251]
lcms(esi ):m/z 680.2(m h);1h-nmr(300mhz,cdcl3):δ9.04(s,1h),7.96(d,j=8.4hz,2h),7.32(s,1h),7.30-7.29(m,6h),7.25(m,2h),5.13-5.09(m,2h),4.80-4.79(m,1h),4.74-4.72(m,1h),3.78-3.73(m,1h),3.60-3.54(m,2h),3.34-3.30(m,2h),2.39(s,3h),1.50(s,9h),1.28-1.25(m,2h),0.96(t,j=5.4hz,3h).
[0252]
步骤5:
[0253][0254]
在氮气保护下,将化合物5(969mg,1.42mmol,1.0eq)溶于无水乙腈(20ml)中依次加入tfa/tfaa(1ml/5ml)。反应液升温回流搅拌反应48小时。冷却至室温后,加入水(40ml)淬灭反应,并用乙酸乙酯(20ml
×
3)萃取,饱和食盐水洗涤合并的有机层,并用无水na2so4干燥。过滤和浓缩后,得到1.4g粗产物,柱层析(dcm/meoh=100/1)纯化,得到509mg化合物6。产率:70%。lcms(esi ):m/z 524.1(m h).
[0255]
步骤6:
[0256][0257]
将化合物6(690mg,1.32mmol,1.0eq)溶于甲醇(10ml)中,降温至0℃,缓慢加入naoh(104mg,2.6mmol,2.0eq)的3ml水溶液。然后反应自然升温至室温并搅拌40分钟。lc-ms跟踪,原料6消耗完。体系加入5ml水淬灭反应,并用6m hcl酸化至ph=2-3。浓缩除去大部分meoh,残留物水层用etoac(3ml
×
3)洗涤。收集水层并浓缩除水,将残留物溶于10ml甲醇中,过滤除去盐,浓缩滤液得到350mg化合物7。lcms(esi ):m/z 274.1(m h)。注意:产品中残留杂质tos-oh。
[0258]
步骤7:
[0259][0260]
在氮气保护下,cdi(207mg,1.28mmol,1eq)溶于dmf(2ml)中并降温至0℃。缓慢滴加a(127mg,1.28mmol,1eq)的dmf(2ml)溶液,加完保温0℃反应40min。另外,化合物7(350mg,1.28mmol,1eq)溶于dmf(2ml)中并加入tea(389mg,3.84mmol,3eq)。然后将含有化合物7的溶液缓慢滴加入到含有化合物a溶液中。加完体系保温0℃反应2小时。lc-ms跟踪,原料7消耗完全。用25ml水淬灭反应,乙酸乙酯(20ml
×
3)萃取,食盐水(20ml
×
2)洗涤合并的有机层,并用无水na2so4干燥。过滤和浓缩后,得到355mg粗产物,通过制备液相进行纯化,然后薄层色谱法(tlc)再次纯化,得到50mg白色固体形式的f粗品。手性hplc分析显示有两组峰。经sfc hplc纯化后,得到20mg产物f(3s,4r)-3-乙基-4-(3h-5-氟基咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺,及其6mg非对映异构体化合物f2(3s,4s)-3-乙基-4-(3h-5-氟基咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺)。
[0261]
化合物f:lcms(esi ):m/z 399.1(m h);1h-nmr(300mhz,cdcl3):δ9.85(br s,1h),8.71(s,1h),7.55(s,1h),7.13(s,1h),4.73(t,j=6.3hz,1h),4.40(m,1h),3.98-3.89(m,4h),3.72-3.69(m,1h),3.49-3.31(m,1h),2.87-2.64(m,1h),1.37-1.25(m,2h),0.79-0.71(m,3h).
[0262]
化合物f2:1h-nmr(300mhz,cdcl3):δ9.87(br s,1h),8.68(s,1h),7.65(s,1h),7.10(s,1h),4.62(t,1h),4.09-4.00(m,1h),3.97-3.85(m,3h),3.44(m,1h),3.30-3.24(m,1h),2.60(s,1h),2.02-2.00(m,1h),1.48-1.37(m,2h),0.97(t,j=7.2hz,3h).
[0263]
实施例5
[0264]
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(3,3,3-三氟丙基)吡咯烷-1-酰胺(g),其制备流程如下:
[0265][0266]
在氮气保护下,cdi(259mg,1.6mmol,1.0eq)溶于无水dmf(2ml)中,并降温至0℃。
缓慢向体系滴加化合物a(239mg,1.6mmol,1.0eq)的0.5ml dmf溶液,加完继续保温搅拌1.5小时。在氮气保护下,将粗品11(400mg,0.3eq)溶于dmf(2ml)中并加入tea(586mg,6.4mmol,4eq)。将含有化合物int-11溶液逐滴加入含有(3,3,3-三氟丙基)氨溶液中,反应温度不超过10℃。加完,反应温度缓慢升至室温反应3小时。tlc跟踪,原料11消耗完全。用20ml冰水淬灭反应,dcm(10ml
×
3)萃取,食盐水(20ml
×
2)洗涤合并的有机层,并用无水na2so4干燥。过滤和浓缩后,得到180mg粗产物,柱层析(dcm/meoh=100:1)纯化,得到53mg灰白色固体g。
[0267]
lcms(esi ):m/z 395.2(m h);hplc:100%;1h-nmr(300mhz,meod):δ8.55(s,1h),7.49(s,1h),7.42(d,j=3.3hz,1h),6.99(d,j=3.6hz,2h),4.43(m,1h),3.90-3.81(m,2h),3.74-3.68(m,1h),3.47(t,j=6.9hz,2h),3.34-3.33(m,2h),2.70(m,1h),2.49-2.40(m,2h),1.32-1.28(m,1h),0.85(m,1h),0.74(t,j=7.2hz3h).
[0268]
实施例6
[0269]
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2-二氟丙氰基)吡咯烷-1-酰胺(h),其制备流程如下(如图5所示):
[0270]
步骤1:
[0271][0272]
将化合物h1(6g,50mmol,1eq)溶解于meoh(25ml)中,然后分批加入试剂hs1(10.57g,55mmol,1.1eq),搅拌分散。然后向上述体系中缓慢滴加甲醛溶液(4.9ml),加完搅拌0.5h。向上述溶液中加入乙醚(20ml),并加热回流过夜。次日,反应生成大量白色固体。lc-ms跟踪,原料h1消耗完全。用水(100ml)淬灭反应并过滤,50ml meoh洗涤滤饼。真空干燥后,得到14.0克白色固体化合物h2。产率:85%。
[0273]
lcms(esi ):m/z 329.2(m h);1h-nmr(300mhz,cdcl3):δ8.08(d,j=3.0hz,1h),7.18-7.98(m,13h),5.54(s,2h),3.78(s,4h).
[0274]
步骤2:
[0275][0276]
在氮气保护下,将锌粉(4.7g,73mmol,3eq)悬浮于thf(70ml)中,依次加入tmscl(4.0g,36.5mmol,1.5eq)以及试剂hs2(4.9g,24.3mmol,1eq)。将化合物h-2(8g,24.3mmol,1eq)的thf(35ml)溶液缓慢滴加至上述体系中。加完室温搅拌2小时。tlc跟踪,原料h-2消耗
完全。用100ml碳酸氢钠溶液淬灭反应,乙酸乙酯(150ml
×
3)萃取,食盐水(100ml
×
2)洗涤合并的有机层,并用无水na2so4干燥。过滤和浓缩后,粗产物通过柱层析(pe/ea=50:1)纯化,得到3.8g化合物h-3。产率:47%。
[0277]
lcms(esi ):m/z 334.1(m h);1h-nmr(300mhz,cdcl3):δ7.22-7.54(m,10h),4.15(q,j=14.4,7.2hz,2h,),3.67(s,3h),3.14(t,j=13.2hz,3h),1.20(t,j=7.2hz,3h).
[0278]
步骤3:
[0279][0280]
化合物h-3(1.7g,5.1mmol,1.0eq)溶于meoh(50ml)溶液中,缓慢加入氨水(15ml),室温搅拌2小时。tlc跟踪,原料h-3消耗完全,直接减压浓缩除去反应溶剂,剩余物用pe/ea=30:1打浆。过滤后,得到1.3g化合物h-4,白色固体。产率:84%。
[0281]
lcms(esi ):m/z 305.1(m h);1h-nmr(300mhz,cdcl3):δ7.23-7.58(m,10h),6.46(br s,1h),6.19(br s,1h),3.96-3.89(m,4h),3.20(t,j=13.5hz,2h).
[0282]
步骤4:
[0283][0284]
化合物h-4(700mg,2.3mmol,1.0eq)溶于meoh(35ml)溶液中,加入pd/c(300mg),氢气置换三次,于氢气氛围下室温搅拌4小时。tlc跟踪,原料h-4消耗完全,过滤浓缩,得到268mg化合物h-5。产率:94%。
[0285]
lcms(esi ):m/z 125.0(m h);1h-nmr(300mhz,cdcl3):δ6.57(brs,1h),5.95(brs,1h),3.26(t,j=14.1hz,2h,),1.33(brs,2h).
[0286]
步骤5:
[0287][0288]
在氮气保护下,cdi(64.8mg,0.4mmol,1.0eq)溶于dmf(2.5ml)中,冰水浴下,体系降温至10℃,缓慢向体系滴加化合物5(49.6mg,0.4mmol,1.0eq)的0.5ml dmf溶液,反应搅拌0.5小时。同时氮气保护下,int-11(120mg,0.36mmol,0.9eq)溶于dmf(2ml)并加入tea
(586mg,6.4mmol,4eq)。将含有化合物int-11溶液滴入含有化合物5的溶液中,然后反应体系自然恢复室温反应1小时。tlc跟踪,原料int-11消耗完全。反应加15ml水淬灭,dcm(15ml
×
3)萃取,食盐水(10ml
×
2)洗涤合并的有机层,并用无水na2so4干燥。过滤和浓缩后干燥浓缩残留物通过硅胶柱(dcm/meoh=50:1~30:1)纯化,得到30mg的灰白色固体化合物h-6。产率:89%。
[0289]
lcms(esi ):m/z 406.2(m h);1h-nmr(300mhz,meod):δ8.55(s,1h),7.50(s,1h),7.42(d,j=5.7hz,1h),7.37(s,1h),7.00(d,j=3.6hz,1h),4.43(m,1h),3.95-3.71(m,4h),3.21(m,1h),2.72(m,1h),1.31(m,1h),0.85(m,1h),0.78-0.74(m,3h).
[0290]
步骤6:
[0291][0292]
在氮气保护下,化合物h-6(60mg,0.148mmol,1.0eq)溶于无水thf(5ml)中,缓慢加入tea(0.13ml),冰水浴降温至10℃左右。向溶液中缓慢滴加tfaa(0.2ml),然后保温反应20min。tlc跟踪,原料h-6消耗完全。体系加入30ml碳酸氢钠水溶液淬灭反应,并用thf:ea=1:1(10ml
×
3)萃取,食盐水(10ml
×
2)洗涤合并的有机层,并用无水na2so4干燥。过滤和浓缩后,残留物通过硅胶柱(dcm/meoh=100/0~20/1)纯化,得到的较纯品用乙醚打浆过滤,得到20mg白色固体目标产物h。
[0293]
lcms(esi ):m/z 388.1(m h);1h-nmr(300mhz,meod):δ8.55(s,1h),7.55(s,1h),7.42(d,j=3.3hz,1h),7.37(s,1h),7.00(d,j=3.6hz,1h),4.43(m,1h),3.95-3.71(m,6h),3.21(m,1h),2.72(m,1h),1.31(m,1h),0.85(m,1h),0.76(t,j=6.9hz,3h).
[0294]
实施例7
[0295]
本实施例提供具有janus激酶抑制活性的化合物(3s,4s)-3-氟基甲基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-酰胺(c2),其制备流程如下:
[0296]
基本按实施例1中所述的方法制备,不同点在于,用中间体trans-9替换实施例1中的中间体cis-9,从而制得目标化合物c2。经检测产物的纯度》97%;lcms(esi ):m/z 384.7(m h).
[0297]
实施例8
[0298]
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-氟基甲基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(3,3,3-三氟丙基)吡咯烷-1-酰胺(a),其制备流程如下:
[0299]
基本按实施例1中所述的方法制备,不同点在于,用(3,3,3-三氟丙基)氨溶液替换
实施例1中的化合物(2,2,2-三氟乙基)氨溶液(试剂s6),从而制得目标化合物a。经检测产物a的纯度》97%;lcms(esi ):m/z398.8(m h).
[0300]
实施例9
[0301]
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(1,1,2,2-四氘-3,3,3-三氟丙基)吡咯烷-1-酰胺(g-4d),其制备流程如下:
[0302]
基本按实施例5中所述的方法制备,不同点在于,用(1,1-二氘-2,2,2-三氟乙基)氨溶液替换实施例5中的化合物(3,3,3-三氟丙基)氨溶液,从而制得目标化合物g-4d。经检测产物的纯度》97%;lcms(esi ):m/z 399.4(m h).
[0303]
实施例10
[0304]
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(1,1-二氘-2,2,2-三氟乙基)吡咯烷-1-酰胺(b),其制备流程如下:
[0305]
基本按实施例5中所述的方法制备,不同点在于,用(1,1-二氘-2,2,2-三氟乙基)氨溶液替换实施例5中的化合物(3,3,3-三氟丙基)氨溶液,从而制得目标化合物b。经检测产物的纯度》97%;lcms(esi ):m/z 382.4(m h).
[0306]
实施例11
[0307]
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(1,1,-二氘-2,2-二氟丙氰基)吡咯烷-1-酰胺(h-2d),其制备流程如下:
[0308]
基本按实施例6中所述的方法制备,不同点在于,用氘代甲醛溶液替换实施例5中的化合物甲醛溶液,从而制得目标化合物h-2d。经检测产物的纯度》97%;lcms(esi ):m/z 390.2(m h).
[0309]
实施例12
[0310]
本实施例提供具有janus激酶抑制活性的化合物(3s,4r)-3-丙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e]吡嗪-8-基)-n-(2,2,-二氟乙基)吡咯烷-1-酰胺(i),其制备流程如下:
[0311]
基本按实施例3中所述的方法制备,不同点在于,用(2,2,-二氟乙基)氨溶液替换实施例3中的化合物(2,2,2-三氟乙基)氨溶液(试剂s6),从而制得目标化合物i。经检测产物的纯度》97%;lcms(esi ):m/z 376.4(m h).
[0312]
验证实施例
[0313]
1.体外jakl激酶活性实验:分别将药物用dmso溶解,配制终浓度为1m的溶液备用;将这些浓度为1m的药物溶液采用梯度稀释法,分别稀释为:1mm、100μm、10μm、1μm、100nm、10nm、1nm,混匀备用;
[0314]
1)配制浓度为1m的dtt溶液备用(溶剂选择dmso);
[0315]
2)jak1试剂盒组分配制(表1)
[0316]
a、将组分中5
×
kinase buffer 1、atp和10
×
irs1-tide置于冰上融化,用5
×
kinase buffer1将浓度为1m的dtt溶液稀释为10mm(如:取10μl 1m dtt溶液加入到1ml5
×
kinase buffer 1混匀);
[0317]
b、试剂盒mix制备:原则:n孔
×
(6μl5
×
kinase buffer 1 1μlatp(500μm) 5μl 10
×
irs1-tide 13μl ddh2o),每孔加入25μl mix。注意:为防止加液过程当中损失,配制mix时,会按照n 1孔的原则来配制;
[0318]
c、每个药物浓度设置3组:阳性对照组、试验组和空白对照。阳性对照组和空白对照组每孔加入5μl inhibitor buffer(no inhibitor),试验组加入5μl对应浓度的待测试药物溶液;
[0319]
d、吸取600μl 5
×
kinase buffer 1,加入2400μl ddh2o,制备出3ml 1
×
kinase buffer 1备用;
[0320]
e、向每个空白对照组的孔中加入20μl 1
×
kinase buffer 1;
[0321]
f、将jak1放在冰上融化并稀释。首次使用时,计算出所需jak1的量,用1
×
kinase buffer 1稀释至终浓度为5ng/μl。将剩余保存至-80℃冰箱;
[0322]
g、向每一个阳性对照组和试验组孔中加入20μl浓度为5ng/μl的jak1,震荡混匀30℃孵育45min;
[0323]
h、将kinase-glo max置于冰上融化备用;
[0324]
i、孵育45min后,向每孔中加入50μl kinase-glo max,震荡混匀后用铝箔将板覆盖,室温反应15min;
[0325]
j、放入酶标仪中读取结果。
[0326]
表1各个实验组的试剂组成成分
[0327] 阳性对照组试验组空白对照组5
×
kinase buffer 16μl6μl6μlatp(500um)1μl1μl1μl10
×
irs1-tide5μl5μl5μlddh2o13μl13μl13μltest inhibitor(待测药物)-5μl-inhibitor buffer(no inhibitor5μl 5μl1
×
kinase buffer 1
‑‑
20μljak1(5ng/μl)20μl20μl-total50μl50μl50μl
[0328]
计算ic50值,结果如下表2。
[0329]
表2计算的ic50值
[0330]
[0331][0332]
2.白癜风实验
[0333]
c57bl小黑鼠,级别spf,体质量(18
±
2)g,雌性。对小鼠进行皮肤表皮脱色按照文献rebecca l.riding,et al.mouse model for human vitiligo.current protocols in immunology,2018进行操作。选择尾部脱色率至少为50%的小鼠进行脱色实验。开始治疗前拍摄所有小鼠的腹侧和背尾照片以确定基线脱色。按剂量分别灌胃给药,给药的容积为20ml
·
kg-1
,每天1次,连续灌胃50天。复色观察选取造模成功动物开始灌胃给药,每10天对实验动物进行摄像评估,观察造模部位色素沉着情况,实验结束后观察造模部位肤色变化并评分评价整体的治疗有效率。
[0334]
由结果可知,化合物c、化合物d、化合物e、化合物f、化合物g和化合物h整体的治疗有效率50-75%。
[0335]
3.药代动力学评价
[0336]
取上述实施例制备获得的化合物,对其进行药代动力学实验,pbs作为空白对照,upadacitinib作为阳性对照。
[0337]
受试动物为:雄性cd1小鼠,体重18-22g,受试动物在试验日前3-7天应在试验场所进行适应性饲养,随机分成2组,分别灌胃给予被试样品,试验前禁食12小时,自由饮水。给药2小时后统一进食。单次灌胃给予25mg/kg剂量的待测样品,待测样品均用0.5%w/v hpmc。
[0338]
分别在给药后0.25、0.5、1.0、2.0、3.0、5.0、8.0、10和24h取样;每个时间点3只小鼠,在以上设定时间点动物麻醉后经小鼠眼球后静脉丛取静脉血0.3ml,置肝素化试管中,11000g离心5分钟,分离血浆,于-20℃冰箱中冷冻。样品检测时,血浆样品经甲醇沉淀蛋白后采用lc-ms/ms法测定血浆中实施例化合物的浓度。
[0339]
采用winnonlin 6.3软件计算小鼠灌胃给药后的主要药动学参数。
[0340]
化合物b相比upadacitinib、化合物g-4d相比化合物g、化合物h-2d相比化合物h的药物在体内的代谢到一半时所需要的时间(半衰期,t
1/2
),分别延长了18%,16%和23%,auc
0-∞
提高了10%-30%。
[0341]
本发明实施例中提供的氘代化合物大大增加了药物的半衰期,延长了药物在人体体内滞留的时间;同时提高血液中药物的浓度,从而达到更好的疗效。
[0342]
以上对本发明的具体实施例进行了详细描述,但其只作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
[0343]
参考文献
[0344]
1.kontzias a,kotlyar a,laurence a,changelian p,o'shea jj.jakinibs:a new class of kinase inhibitors in cancer and autoimmune disease.current opinion in pharmacology,2012,12(4):464
–
470.
[0345]
2.pesu m,laurence a,kishore n,zwillich sh,chan g,o'shea jj.therapeutic targeting of janus kinases.immunological reviews,2008,223:132
–
42.
[0346]
3.norman p.selective jak inhibitors in development for rheumatoid arthritis.expert opinion on investigational drugs.2014,23(8):1067
–
77.
[0347]
4.jak inhibitors showing promise for many skin problems-conditions ranging from alopecia to vitiligo.2017.
[0348]
5.perris,a.b.and p.b.rothman.jak-stat signaling in asthma.the journal of clinical investigation,2002,109(10):1279-83.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。