一种sirt2/hdac6双靶标抑制剂及其应用
技术领域
1.本发明涉及化学技术和药学技术领域,尤其是一种一种sirt2/hdac6双靶标抑制剂及其应用。
背景技术:
2.微管(microtubule)是细胞骨架的主要组成部分,同时,它的动态组装在细胞有丝分裂和细胞增殖过程中具有独特的作用,是目前一线抗肿瘤药物如紫杉醇、长春碱和秋水仙碱等的作用靶点。微管(microtubule)也是第一个被鉴定为非组蛋白的乙酰化底物,微管(microtubule)是由微管蛋白α-tubulin和β-tubulin形成的二聚体组装而成的,其中α-tubulin的第40个赖氨酸(k40)被证实是其乙酰化位点。研究表明,α-tubulin k40乙酰化水平可以作为稳定微管、调节微管结构和控制应激和免疫反应的标记物;此外,k40乙酰化水平在其他各种细胞过程中具有很重要的潜在作用,包括细胞内的运输,纤毛组装、细胞信号转导、细胞迁移等。
3.研究发现α-tubulin k40乙酰化水平能被微管蛋白乙酰化转移酶(alpha-tubulin acetyltransferase 1,ata1)和组蛋白去乙酰化酶家族成员hdac和sirtuin调控。其中,hdac6是第一个被证实的能够去除α-tubulin k40乙酰化的组蛋白去乙酰化酶,随后,sirt2也被发现能够去除α-tubulin k40乙酰化。虽然hdac6是主要的α-tubulin k40去乙酰化酶,但是,sirt2则在某些条件下代偿性地α-tubulin k40去乙酰化酶,例如,在细胞有丝分裂纺锤体阶段,主要是sirt2下调α-tubulin k40乙酰水平;在巨噬细胞炎性体激活阶段,主要是sirt2表现出α-tubulin k40去乙酰化酶活性。更为重要的是,在多种病理学上,微管蛋白α-tubulin k40乙酰化水平大多是呈现显著降低情况,比如:阿尔茨海默病、帕金森病等神经系统疾病;多发性骨髓瘤、圆柱瘤等肿瘤疾病;心房纤颤等心脏病;慢性阻塞性肺疾病(copd);结肠炎、移植排斥等炎症和免疫疾病等等。由此可见,α-tubulin的k40乙酰化水平降低与肿瘤、神经性退行疾病、心脏疾病、炎症、病毒感染等疾病密切相关。
4.ras是经典的肿瘤治疗通路中发挥至关重要的一个蛋白,egfr通过激活ras蛋白,调控其下游raf与pi3k等蛋白的磷酸化,在通过级联放大调控多种肿瘤基因的表达。k-ras是ras蛋白的一种突变体,这种突变在胰腺癌、非小细胞肺癌等多种癌症中占有很高比例,且该突变体对靶向egfr的药物表现出很强得耐受性,因此k-ras也成为现今肿瘤治疗非常重要的靶点。2013年yang等人发现sirt2可通过下调k-ras第104位赖氨酸的乙酰化水平而上调其下游底物的磷酸化水平,从而促进肿瘤的增殖与迁移,在对其机制进行研究的时候,研究人员发现,当给予sirt2抑制剂时,该通路并不会被抑制,后续发现当sirt2活性下降时,细胞可通过代偿提高hdac6的活性以降低k-ras k104的乙酰化水平。因此,能够下调α-tubulin k40和k-ras k104乙酰化水平的hdac6和sirt2已成为相关疾病药物研发的热点靶标,hdac6抑制剂和sirt2抑制剂也是相关疾病药物研发的研究前沿和热点。因此,对于hdac6和sirt2双靶标抑制剂的研究与开发具有非常重要的理论和现实意义。然而,目前仅有关于hdac与其他靶标的双抑制剂的研究报道,比如,hdac和fgfr1、hdac与pi3k、hdac与
pde5、hdac与ras、hdac与jak2、hdac与topoisomerase ii、hdac与lsd1、hdac与nampt等双抑制剂。对于hdac与sirtuin,特别是高选择性的hdac6与sirt2双靶点抑制剂均为见报道。
技术实现要素:
5.本发明的目的是:提供一种sirt2/hdac6双靶标抑制剂及其应用,它在体内外能够有效的选择性的抑制sirt2和hdac6的活性,且本发明合成原料便宜,成本较低,对药物筛选和制药行业具有重要的应用价值。
6.本发明是这样实现的:一种sirt2/hdac6双靶标抑制剂,该化合物以赖氨酸为骨架,并具有如下通式之一所示的结构:
[0007][0008][0009]
式中,所述r1为亚甲基或亚氨基;r2为十四烷基、苯基或金刚烷基、聚乙氧基;r3为烷基、苯基、苯甲基、环己烷基、聚乙氧基、苯并噻唑基、噻唑基或哌嗪基;r4为异羟肟酸、巯基、邻苯二胺基、对氟邻苯二胺基或乙基乙酰肼基;r5为喹啉基、苯甲基、金刚烷基、三苯胺基、吡啶并[4,3-b]吲哚基、1,2,3,4-四氢喹啉基、吲哚基、苯并噻唑基、苯并噻吩基或5-苯基异恶唑基;r6为烷基、苯基、聚乙氧基或哌嗪基。
[0010]
其合成路线为以下四种合成路线之一:
[0011]
合成路线1:
[0012][0013]
步骤a:将甘氨酸(5mmol,1eq)溶于无水甲醇中,缓慢滴加氯化亚砜(5mmol,1eq),
然后回流2h,减压干燥,得到产物a。
[0014]
步骤b:将硫代cbz-lys(1mmol,1eq)溶于无水四氢呋喃中,然后加入diea(3mmol,3eq),再加入hbtu(1.5mmol,1.5eq),室温搅拌10min后加入产物a,室温反应4h后减压除去溶剂,残渣用二氯甲烷溶解,饱和氯化钠洗涤三次,无水硫酸钠干燥,pe/ea=2:1柱层析得到产物b。
[0015]
步骤c:将产物b(1mmol,1eq)溶于无水甲醇中,然后加入盐酸羟胺(20mmol,20eq),再加入氢氧化钾(22mmol,22eq),室温反应4h后减压除去溶剂,dcm/meoh=20:1柱层析得到产物c。
[0016]
合成路线2:
[0017][0018]
步骤d:将硫代cbz-lys(1mmol,1eq)溶于无水四氢呋喃中,然后加入diea(3mmol,3eq),再加入hbtu(1.5mmol,1.5eq),室温搅拌10min后加入5-苯基异恶唑胺(1.2mmol,1.2eq),室温反应4h后减压除去溶剂,残渣用二氯甲烷溶解,饱和氯化钠洗涤三次,无水硫酸钠干燥,pe:ea=1:1柱层析得到产物d。
[0019]
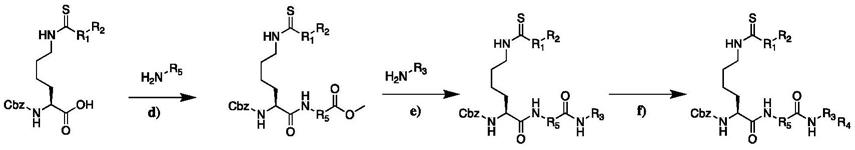
步骤e:将产物d(1mmol,1eq)溶于thf/h2o=1:1中,然后加入氢氧化锂(3mmol,3eq),过夜反应后调节ph至3,ea萃取,浓缩干燥后溶于无水thf中,然后加入diea(3mmol,3eq),再加入hbtu(1.5mmol,1.5eq),室温搅拌10min后加入7-氨基庚酸甲酯(1.2mmol,1.2eq),室温反应4h后减压除去溶剂,残渣用二氯甲烷溶解,饱和氯化钠洗涤三次,无水硫酸钠干燥,pe:ea=1:2柱层析得到产物e。
[0020]
步骤f:将产物e(1mmol,1eq)溶于无水甲醇中,然后加入盐酸羟胺(20mmol,20eq),再加入氢氧化钾(22mmol,22eq),室温反应4h后减压除去溶剂,dcm/meoh=20:1柱层析得到产物f。
[0021]
合成路线3:
[0022][0023]
步骤g:将硫代cbz-lys(1mmol,1eq)溶于无水四氢呋喃中,然后加入diea(3mmol,3eq),再加入hbtu(1.5mmol,1.5eq),室温搅拌10min后加入4-叠氮基苯胺,室温反应4h后减压除去溶剂,残渣用二氯甲烷溶解,饱和氯化钠洗涤三次,无水硫酸钠干燥,pe/ea=2:1柱层析得到产物g。
[0024]
步骤h:将产物g(0.1mmol,1eq)溶于无水dmf中,然后加入n-hydroxy-4-((prop-2-yn-1-yl(quinolin-8-yl)amino)methyl)benzamide(0.1mmol,1eq),然后加入无水硫酸铜(0.02mmol,0.2eq),再加入tbta(0.01mmol,0.1eq),然后将抗坏血酸钠(0.05mmol,0.5eq)
溶于200μl水中,室温反应16h后加入ea,饱和氯化钠洗涤三次,无水硫酸钠干燥,dcm/meoh=10:1柱层析得到产物h。
[0025]
合成路线4:
[0026][0027]
步骤g:将硫代cbz-lys(1mmol,1eq)溶于无水四氢呋喃中,然后加入diea(3mmol,3eq),再加入hbtu(1.5mmol,1.5eq),室温搅拌10min后加入4-叠氮基苯胺,室温反应4h后减压除去溶剂,残渣用二氯甲烷溶解,饱和氯化钠洗涤三次,无水硫酸钠干燥,pe/ea=2:1柱层析得到产物g。
[0028]
步骤h:将产物g(0.1mmol,1eq)溶于无水dmf中,然后再加入(e)-n-hydroxy-4-(((2'-oxo-1'-(prop-2-yn-1-yl)-[2,3'-biindolin]-3-ylidene)amino)oxy)butanamide(0.1mmol,1eq),然后加入无水硫酸铜(0.02mmol,0.2eq),再加入tbta(0.01mmol,0.1eq),然后将抗坏血酸钠(0.05mmol,0.5eq)溶于200μl水中,室温反应16h后加入ea,饱和氯化钠洗涤三次,无水硫酸钠干燥,dcm/meoh=10:1柱层析得到产物i。
[0029]
上述路线中,所述r1为亚甲基或亚氨基;r2为十四烷基、苯基或金刚烷基、聚乙氧基;r3为烷基、苯基、苯甲基、环己烷基、聚乙氧基、苯并噻唑基、噻唑基或哌嗪基;r4为异羟肟酸、巯基、邻苯二胺基、对氟邻苯二胺基或乙基乙酰肼基;r5为喹啉基、苯甲基、金刚烷基、三苯胺基、吡啶并[4,3-b]吲哚基、1,2,3,4-四氢喹啉基、吲哚基、苯并噻唑基、苯并噻吩基或5-苯基异恶唑基;r6为烷基、苯基、聚乙氧基或哌嗪基。
[0030]
sirt2/hdac6双靶标抑制剂制备实体瘤与血液瘤药物中的应用。
[0031]
通过采用上述技术方案,本发明的sirt2/hdac6双靶标抑制剂在体内外能够有效的选择性的抑制sirt2和hdac6的活性,且本发明合成原料便宜,成本较低,抗肿瘤活性明显,可用于高效低毒的新型sirt2/hdac6双靶标抑制剂类抗肿瘤药物。
具体实施方式
[0032]
本发明的实施例:采用以下(1)sirt2/hdac6双靶标抑制剂的合成路线:
[0033][0034]
通过该合成路线,获得了化合物1-3,化合物1-3的结构如下:
[0035][0036]
本发明的实施例:采用以下(2)sirt2/hdac6双靶标抑制剂的合成路线:
[0037][0038]
通过该合成路线,获得了化合物4,化合物4的结构如下:
[0039][0040]
本发明的实施例:采用以下(3)sirt2/hdac6双靶标抑制剂的合成路线:
[0041][0042]
通过该合成路线,获得了化合物5,化合物5的结构如下:
[0043][0044]
本发明的实施例:采用以下(4)sirt2/hdac6双靶标抑制剂的合成路线:
[0045]
通过该合成路线,获得了化合物6,化合物6的结构如下:
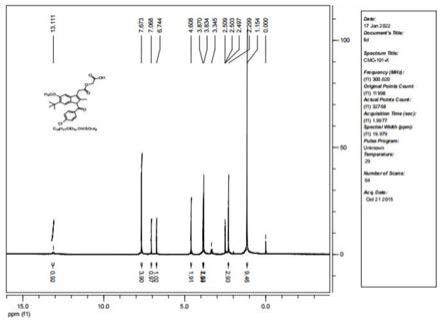
nmr(100mhz,dmso-d6)δ204.51,171.95,171.26,170.38,160.18,159.02,158.22,150.09,141.22,140.29,132.02,131.92,130.19,130.02,129.27,123.15,123.02,120.09,100.97,65.97,60.50,45.27,43.37,42.67,40.73,38.15,36.58,35.19,34.26,31.15,30.58,30.49,29.26,23.16,23.10,18.93,14.22.hrms(esi)for c
45h66
n4o7s(m h
):calcd,835.47865.,found,835.47802.
[0052]
化合物5:1h nmr(400mhz,dmso-d6)δ(ppm):10.51(d,j=5.2hz,2h),8.40(t,j=4.9hz,2h),7.81(d,j=6.2hz,4h),7.72(d,j=6.1hz,4h),7.61(d,j=2.4hz,4h),7.52(d,j=4.1hz,4h),7.32(dd,j=5.2hz,4h),7.02(d,j=2.2hz,1h),5.62(d,j=5.2hz,2h),5.02(t,j=4.2hz,2h),4.22(t,j=5.5hz,1h),3.92(t,j=6.1hz,2h),3.80(t,j=4.3hz,2h),3.52(t,j=2.3hz,4h),3.22(t,j=6.3hz,2h),2.97(d,j=11.7,6.7hz,6h),2.48(dd,j=4.2hz,2h),1.72(t,6h),1.65
–
1.58(m,4h),1.49(dt,j=15.0,7.4hz,4h),1.40
–
1.09(m,22h),0.96(t,j=7.0hz,3h).
13
c nmr(100mhz,dmso-d6)δ204.21,173.25,171.06,162.48,157.82,142.32,142.12,141.09,140.42,139.52,130.09,129.87,129.02,128.81,127.69,122.19,120.32,101.27,100.15,67.28,65.42,64.29,60.09,58.29,56.72,54.29,53.19,50.09,43.17,42.07,41.73,37.05,35.28,35.09,33.26,31.05,30.18,30.09,29.23,23.12,23.19,18.24,14.37.hrms(esi)for c
61h80n10
o7s(m h
):calcd,1097.60049.,found,1097.60012.
[0053]
化合物6:1h nmr(400mhz,dmso-d6)δ(ppm):10.57(d,j=5.2hz,2h),8.50(t,j=4.9hz,2h),7.83(d,j=6.2hz,4h),7.62(d,j=6.1hz,4h),7.54(d,j=2.6hz,4h),7.52(d,j=4.1hz,2h),7.02(d,j=4.2hz,2h),6.92(d,j=2.1hz,2h),5.05(t,j=4.1hz,2h),4.85(t,j=6.1hz,2h),4.29(t,j=5.1hz,1h),3.91(t,j=6.1hz,2h),3.86(t,j=4.1hz,2h),3.42(t,j=5.3hz,4h),3.22(t,j=6.3hz,2h),3.02(d,j=11.2,6.7hz,2h),2.49(dd,j=4.1hz,2h),2.29(d,j=6.1hz,2h),1.71(t,6h),1.62
–
1.59(m,4h),1.44(dt,j=13.0,7.1hz,4h),1.40
–
1.09(m,22h),0.91(t,j=6.2hz,3h).
13
c nmr(100mhz,dmso-d6)δ204.41,173.15,172.06,171.09,170.92,169.99,155.48,147.82,142.12,141.12,131.29,130.82,130.52,130.29,129.77,129.02,128.91,127.69,122.19,120.22,111.28,110.25,109.87,71.09,70.97,67.28,61.42,64.29,60.09,52.12,50.09,44.27,42.42,41.33,36.15,35.08,35.29,33.16,31.23,30.28,30.02,29.23,23.22,23.13,18.24,14.58.hrms(esi)for c
62h79
n11o9s(m h
):calcd,1154.58557.,found,1154.58237.
[0054]
实施例2:
[0055]
体外sirt2与hdac6抑制活性筛选:
[0056]
sirt2抑制活性筛选:
[0057]
具体的操作步骤如下:
[0058]
1)按照表1配置体外抑制活性的酶促反应混合液(ddwater的反应体积为38.4μl),将所需测定样品的反应液配置于1.5ml的离心管中;
[0059]
2)将酶促反应混合液分装于标记好的1.5ml离心管中(52μl/个),加入2μl 6μmol/l目标化合物(终浓度0.2μmol/l),加完放于冰盒中;
[0060]
3)涡旋,离心,将全部样品一起放于孵育箱中,37℃预孵育30min;
[0061]
4)取出样品,置于冰盒中放冷,依次加入6μl的底物(终浓度:200μm),总反应体积
为60μl;
[0062]
5)涡旋,离心,30s间隔加一个样品,37℃预孵育60min,依次用quench buffer淬灭液进行淬灭反应;
[0063]
6)10000rpm高速离心10min,取样100μl于96孔板中,在hplc的紫外280nm波长处进行检测,进样体积为40μl。
[0064]
实验中设立1个平行对照组,一个阴性对照(dmso),一个阳性对照tm(终浓度0.2μm)。
[0065]
表1 sirtuin体外抑制活性的酶促反应混合液组成
[0066][0067]
hplc液相色谱检测条件如下:
[0068]
1)色谱柱:inertsustain aq-c18 250
×
46mm,5μm;
[0069]
2)流动相:a相,0.1%tfa/乙腈(v/v);b相,0.1%tfa/ddwater(v/v);
[0070]
3)检测波长:254nm和284nm;
[0071]
4)柱温:25℃
[0072]
5)洗脱条件如下:
[0073]
表2 hplc洗脱梯度条件
[0074][0075]
底物的转化率计算公式见1.1:
[0076]
conversion%=a产物/(a产物 a底物)*100% (1.1)
[0077]
目标化合物的抑制率计算公式见1.2:
[0078]
inhibition%=(1-c目标化合物/c阴性对照)*100% (1.2)
[0079]
化合物1-3的sirt2抑制活性结果如表3:
[0080]
表3化合物1-6的sirt2抑制活性
[0081][0082]
hdac6抑制活性筛选:
[0083]
所有hdac试剂盒均购自bps bioscience。遵循制造商的方案进行酶促hdac测定。以hdac6为例。(荧光hdac6测定试剂盒,目录号:50076;bps bioscience)。
[0084]
具体的操作步骤如下:
[0085]
1)将hdac分析缓冲液(35μl)与bsa(1mg/ml,5μl)和hdac底物(200μm,5μl)在96孔黑色板上混合;
[0086]
2)将hdac6(7ng/μl,5μl)和各种浓度的化合物(5μl)或tsa(5μl)(作为阳性对照)添加到孔中;
[0087]
3)将所得混合物在37℃下孵育30分钟;
[0088]
4)孵育后,将50μl 2xhdac developer添加到每个孔中;
[0089]
5)将混合物在室温下孵育15分钟后,使用酶标仪在360nm激发和460nm发射波长下测量荧光强度。
[0090]
化合物1-6的hdac6抑制活性结果如表4:
[0091]
表4化合物1-6的hdac6抑制活性
[0092][0093]
实施例3:
[0094]
sirt2/hdac6双靶标抑制剂对不同肿瘤细胞的抗肿瘤活性:
[0095]
采用公认的可用于大规模抗肿瘤药物筛选、细胞毒性实验测定的cck8法评价候选化合物对5种人癌细胞的抗增殖活性。测试化合物为化合物3,阴性对照组为不加药组;阳性对照组为tm和acy-1215。
[0096]
细胞株:人乳腺癌细胞mcf-7、人肝癌细胞hepg2、人结直肠癌细胞hct-116、人急性
白血病细胞mv411、人髓性白血病细胞k562。
[0097]
细胞增殖抑制率=(阴性对照组od值-药物组od值)*100%/阴性对照组od值。通过化合物系列浓度的抑制率计算得到ic50值(μm),结果见表5。
[0098]
表5 cck8法测定化合物3对不同肿瘤细胞的ic
50
值(μm)
[0099][0100]
在此有必要指出的是,以上实施例和试验例仅限于对本发明技术方案做进一步阐述和理解,不能理解为对本发明的技术方案做进一步的限定,本领域技术人员作出的非突出实质性特征和显著进步的发明创造,仍然属于本发明的保护范畴。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。