1.本发明涉及一种分离虎杖水提药渣中虎杖苷和大黄素的方法。
背景技术:
2.虎杖为蓼科植物虎杖(polygonum cuspidatum sieb.et zucc.)的干燥根茎和根。微苦,微寒。归肝、胆、肺经。能利湿退黄,清热解毒,散瘀止痛,止咳化痰。用于湿热黄疸,淋浊,带下,风湿痹痛,痈肿疮毒,水火烫伤,经闭,癥瘕,跌打损伤,肺热咳嗽。虎杖苷和大黄素是虎杖的有效成分和指标性成分,不仅对人体具有重要的生理活性,而且是中医药研究和分析测试中常用的化学对照品,因此,制备虎杖苷和大黄素具有重要的应用价值。
3.中药配方颗粒是由单味中药饮片制成的颗粒。中药配方颗粒的制备,除成型工艺外,其余应与传统汤剂基本一致,即以水为溶媒提取,以物理方法固液分离、浓缩、干燥、颗粒成型等工艺生产。中药配方颗粒药效物质应与中药饮片水煎汤剂保持基本一致。中药配方颗粒质量标准规定了出膏率的上下限,在生产过程中,提取率过高会突破这一范围,导致产品不合格。此外,中药配方颗粒以水为溶媒提取,低极性、水溶性差的成分的提取率较低。因此中药配方颗粒提取后的药渣中必然有一定的成分残留。这一方面造成了药材的浪费。另一方面,为防止药渣重新流入市场,企业需对药渣进行销毁处理,增加了工序和成本投入。因此药渣的综合利用是我国发展药材循环经济、建设资源节约型、环境友好型社会的必要一环。目前尚无从虎杖水提药渣中同时分离制备虎杖苷和大黄素的方法报道。
技术实现要素:
4.有鉴于此,本发明提供了一种分离虎杖水提药渣中虎杖苷和大黄素的方法,该方法能够同时将虎杖苷和大黄素从虎杖水提药渣中分离出来。进一步地,该方法工艺简单,可以进而得到高纯度的虎杖苷和大黄素。
5.本发明提供了一种分离虎杖水提药渣中虎杖苷和大黄素的方法,包括如下步骤:
6.将虎杖水提药渣提取物上径高比小于等于1:30的羟丙基葡聚糖凝胶色谱柱,洗脱羟丙基葡聚糖凝胶色谱柱,收集洗脱液,分别得到虎杖苷粗品和大黄素粗品。
7.羟丙基葡聚糖凝胶色谱柱易于与中药复杂组分发生死吸附而变性报废,常用于成分的细分,较少用于提取物的粗分。申请人意外地发现,采用羟丙基葡聚糖凝胶色谱能够同时将虎杖水提药渣提取物中的虎杖苷和大黄素分离出来。
8.本发明中,所述虎杖水提药渣是指将虎杖药材用水提取后得到的药渣,比如生产虎杖配方颗粒时将虎杖药材用水提取后的得到的药渣。
9.在本发明中,羟丙基葡聚糖凝胶色谱柱的径高比小于等于1:30;优选地,羟丙基葡聚糖凝胶色谱柱的径高比为1:50~200。更优选地,羟丙基葡聚糖凝胶色谱柱的径高比为1:90~170。根据本发明的一个实施方式,羟丙基葡聚糖凝胶色谱柱的径高比为1:90~120。这样既能将虎杖水提药渣提取物中的虎杖苷和大黄素完全分离出来,又能够缩短分离时间。
10.根据本发明的方法,优选地,洗脱羟丙基葡聚糖凝胶色谱柱方式为等度洗脱。本发
明采用等度洗脱即可将虎杖水提药渣提取物中的虎杖苷和大黄素完全分离,这样更加易于操作。
11.根据本发明的方法,优选地,洗脱所述羟丙基葡聚糖凝胶色谱柱的流动相选自甲醇、氯仿
‑
甲醇、丙酮、甲醇
‑
水中的一种或多种。氯仿
‑
甲醇中氯仿和甲醇的体积比可以为0.5~2:1;优选为1:1。甲醇
‑
水中甲醇的体积分数可以为50~90vol%;优选为75vol%。根据本发明的一个实施方式,流动相为甲醇。这样能够使虎杖苷和大黄素达到更好的分离度,且溶剂易于回收。
12.根据本发明的方法,优选地,所述流动相的用量为柱体积的3~12倍。更优选地,所述流动相的用量为柱体积的4~7倍。这样既能够将虎杖苷和大黄素组分完全分离,又能够减少流动相的用量,易于回收。
13.根据本发明的方法,优选地,所述羟丙基葡聚糖凝胶色谱柱为sephadex lh
‑
20色谱柱。这样能够使虎杖苷和大黄素组分更好地分离。
14.根据本发明的方法,所述虎杖水提药渣提取物是指将所述虎杖水提药渣用有机溶剂提取后得到的提取物。所述虎杖水提药渣提取物可以采用如下方法制备:
15.采用溶剂提取虎杖水提药渣,得到提取液;将提取液过滤,回收溶剂,得到虎杖水提药渣提取物;
16.其中,所述溶剂选自甲醇、乙醇、60~95vol%的甲醇水溶液、60~95vol%的乙醇水溶液中的一种或多种;所述溶剂的用量为虎杖水提药渣质量的2~20倍。
17.在本发明中,优选地,溶剂为甲醇或乙醇。根据本发明的一个实施方式,溶剂为甲醇,从而将虎杖苷和大黄素充分提取出来,且易于会回收。
18.根据本发明的方法,优选地,所述虎杖水提药渣中虎杖苷的含量为0.045~0.09wt%,所述虎杖水提药渣中大黄素的含量为0.18~0.5wt%。更优选地,所述虎杖水提药渣中虎杖苷的含量为0.05~0.06wt%,所述虎杖水提药渣中大黄素的含量为0.25~0.35wt%。
19.在本发明中,提取方式可以选自浸泡提取、超声提取或加热回流提取中的一种。优选地,提取方式为超声提取或加热回流提取。这样能够得到更高的提取率。
20.加热回流提取或超声提取的提取时间可以为0.4~5h;优选为0.6~1.5h。溶剂的用量可以为虎杖水提药渣质量的2~20倍;优选为4~6倍。
21.浸泡提取的提取时间可以为8~24h;优选为16~24h。溶剂的用量可以为虎杖水提药渣质量的5~20倍;优选为8~12倍。
22.根据本发明的方法,优选地,所述提取虎杖水提药渣的方式选自浸泡提取、超声提取或加热回流提取中的一种,所述提取时间为0.4~24h。
23.在本发明的方法中,还可以包括将虎杖苷粗品和/或大黄素粗品精制的步骤。
24.虎杖苷粗品精制可以包括如下步骤:将虎杖苷粗品采用液相色谱进行分离,收集虎杖苷色谱段的溶液,回收溶剂,得到虎杖苷。
25.根据本发明的一个实施方式,虎杖苷粗品精制的色谱条件为:色谱柱:c
18
色谱柱(9.4mm
×
250mm,5μm);流动相:甲醇a与水b;梯度洗脱程序:0~30min,10vol%~20vol%a,流速2ml/min。
26.根据本发明的另一个实施方式,虎杖苷粗品精制的色谱条件为:色谱柱:c
18
色谱柱
(30mm
×
250mm,10μm);流动相:乙腈a与水b;等度洗脱程序:10vol%a,流速10ml/min。
27.根据本发明的再一个实施方式,虎杖苷粗品精制的色谱条件为:色谱柱:c
18
色谱柱(50mm
×
250mm,10μm);流动相:甲醇a与水b;等度洗脱程序:15vol%a,流速30ml/min。
28.大黄素粗品精制可以包括如下步骤:将大黄素粗品采用液相色谱进行分离,收集大黄素色谱段的溶液,回收溶剂,得到大黄素。
29.根据本发明的一个实施方式,大黄素粗品精制的色谱条件为:色谱柱:c
18
色谱柱(9.4mm
×
250mm,5μm);流动相:甲醇a与水b;梯度洗脱程序:0~30min,70vol%~90vol%a,流速2ml/min。
30.根据本发明的另一个实施方式,大黄素粗品精制的色谱条件为:色谱柱:c
18
色谱柱(30mm
×
250mm,10μm);流动相:乙腈a与水b;等度洗脱程序:60vol%a,流速10ml/min。
31.根据本发明的再一个实施方式,大黄素粗品精制的色谱条件为:色谱柱:c
18
色谱柱(50mm
×
250mm,10μm);流动相:甲醇a与水b;等度洗脱程序:80vol%a,流速30ml/min。
32.根据本发明的方法,优选地,还包括将虎杖苷粗品纯化的步骤:
33.将虎杖苷粗品采用液相色谱进行分离,收集虎杖苷色谱段的溶液,回收溶剂,得到虎杖苷;
34.色谱条件选自如下条件之一:
35.(a1)色谱柱:c
18
色谱柱;流动相:甲醇a与水b;梯度洗脱程序:0~30min,10vol%~20vol%a;
36.(b1)色谱柱:c
18
色谱柱;流动相:乙腈a与水b;等度洗脱程序:10vol%a;
37.(c1)色谱柱:c
18
色谱柱;流动相:甲醇a与水b;等度洗脱程序:15vol%a。
38.根据本发明的方法,优选地,还包括将大黄素粗品纯化的步骤:
39.将大黄素粗品采用液相色谱进行分离,收集大黄素色谱段的溶液,回收溶剂,得到大黄素;
40.色谱条件选自如下条件之一:
41.(a2)色谱柱:c
18
色谱柱;流动相:甲醇a与水b;梯度洗脱程序:0~30min,70vol%~90vol%a;
42.(b2)色谱柱:c
18
色谱柱;流动相:乙腈a与水b;等度洗脱程序:60vol%a;
43.(c2)色谱柱:c
18
色谱柱;流动相:甲醇a与水b;等度洗脱程序:80vol%a。
44.申请人发现,虎杖水提药渣中仍然存在着虎杖苷和大黄素,由于虎杖水提药渣通常直接作为废弃物丢弃,因而这部分虎杖苷和大黄素也被浪费掉了。本发明采用羟丙基葡聚糖凝胶色谱柱能够将虎杖水提药渣中残存的虎杖苷和大黄素同时分离出来,变废为宝,提高了资源利用率,具有环境效益和经济效益。本发明的方法工序简便、步骤少、耗时短、易于控制、分离效率高,溶剂易于回收。进一步地,本发明的方法能够制得高纯度的虎杖苷和大黄素,纯度达到99%以上,可作为标准品使用,或用于质量控制和药理药效研究。本发明的方法稳定性高、重现性好,且易于放大,能够制备克级的虎杖苷和大黄素单体化合物。
具体实施方式
45.下面介绍仪器:
46.agilent 1260高效液相色谱仪,购买自安捷伦科技公司。acquity uplc超高压液
相色谱仪,购买自沃特世公司。
47.b5510e
‑
dth超声提取仪,购买自必能信超声有限公司。
48.下面介绍原料:
49.虎杖水提药渣为康美药业股份有限公司生产虎杖配方颗粒后的药渣,即虎杖用水提取之后的药渣;虎杖苷对照品购买自中国食品药品检定研究院;大黄素对照品购买自中国食品药品检定研究院。
50.色谱溶剂为色谱纯,水为超纯水(由购买自美国millipore公司的milli
‑
q超纯水机制得),其他试剂为分析纯。
51.干膏得率的测定方法:虎杖水提药渣提取物蒸干后,于105℃干燥3小时,置干燥器中冷却30分钟,迅速精密称定重量。以本次提取投料的虎杖水提药渣干燥品重量计算干膏得率。
52.检测虎杖水提药渣中虎杖苷、大黄素的含量
53.分别取10批康美药业股份有限公司生产虎杖配方颗粒后的虎杖水提药渣,标号依次为批次1至批次10。将批次1至批次10的虎杖水提药渣分别制成供试品溶液。供试品溶液的制备方法如下:取虎杖水提药渣适量,研细,取1.0g,置于具塞锥形瓶中,精密加入甲醇50ml,称定重量,超声处理(功率250w,频率40khz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
54.将虎杖苷对照和大黄素对照分别制成对照品溶液。对照品溶液的制备如下:向对照品中加入甲醇制成每1ml含100μg对照品的储备液,混合、稀释得一系列浓度的对照品溶液。
55.色谱条件:色谱柱:acquity beh c18色谱柱(2.1mm
×
100mm,1.7μm);流动相:乙腈a与0.2vol%甲酸溶液b;梯度洗脱:0~7min,12~20vol%a;7~10min,20~28vol%a;10~12min 28vol%a;12~15min 28~30vol%a;15~16min 30~80vol%a;16~18min 80vol%a);流速:0.4ml/min;柱温为40℃;检测波长为290nm。
56.测定法:分别精密吸取对照品溶液与供试品溶液,注入acquity uplc液相色谱仪,测定,即得。
57.测定结果:按干燥品计算,批次1至批次10的虎杖水提药渣中虎杖苷的含量分别为:0.055wt%、0.059wt%、0.061wt%、0.053wt%、0.072wt%、0.081wt%、0.063wt%、0.071wt%、0.069wt%、0.051wt%。按干燥品计算,批次1至批次10的虎杖水提药渣中大黄素的含量分别为:0.31wt%、0.25wt%、0.23wt%、0.22wt%、0.31wt%、0.35wt%、0.28wt%、0.40wt%、0.29wt%、0.27wt%。
58.可见,虎杖水提药渣中仍然含有较高含量的虎杖苷和大黄素。
59.以下以批次1的虎杖水提药渣为原料,提取分离虎杖苷和大黄素。
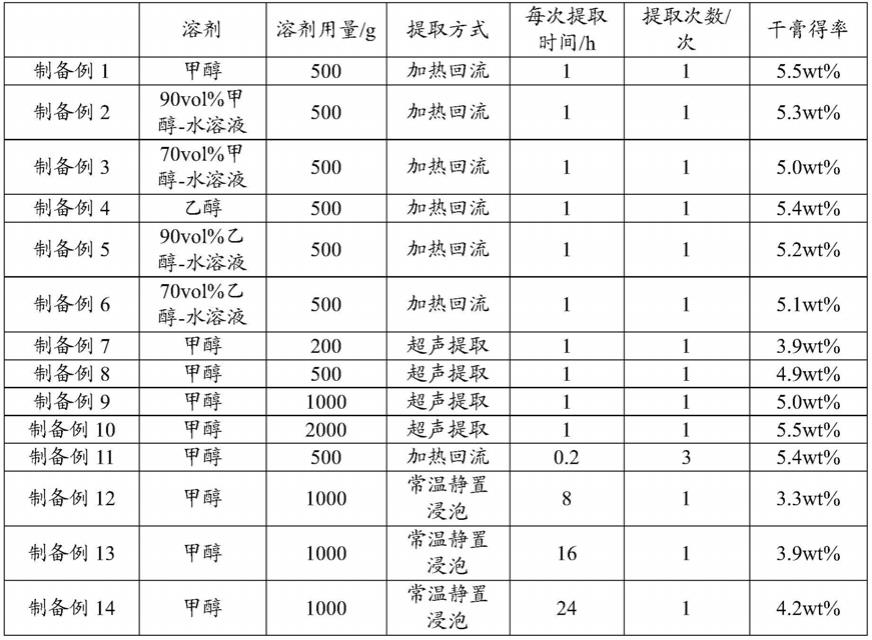
60.制备例1~14
61.取100g虎杖水提药渣,加入溶剂,提取;将提取液过滤,回收溶剂,得到虎杖水提药渣提取物。具体参数如表1所示,干膏得率如表1所示。
62.表1
[0063][0064]
由表1可知,甲醇、甲醇
‑
水溶液、乙醇、乙醇
‑
水溶液均能用于虎杖水提药渣的提取,甲醇的提取效果较好且易于回收,因此选择甲醇为溶剂。干膏得率随着溶剂用量的增加而增加,当溶剂的用量达到虎杖水提药渣质量的5倍后,干膏得率的增长趋于平缓,考虑到后期溶剂回收及成本,溶剂的用量优选为虎杖水提药渣质量的5倍。采用加热回流或超声提取的方式能够获得更高的干膏得率。
[0065]
制备例15
[0066]
取5kg虎杖水提药渣,加入25kg甲醇,加热回流提取1h;将提取液过滤,回收溶剂,得到虎杖水提药渣提取物0.25kg。
[0067]
实施例1~8及对比例1
[0068]
sephadex lh
‑
20色谱柱(sephadex lh
‑
20填料900g),径高比如表2所示,用流动相溶胀sephadex lh
‑
20填料,装成9根每根含填料100g的柱子。取制备例15得到的虎杖水提药渣提取物18g,将虎杖水提药渣提取物溶解于流动相中,上样,每根柱子上样量均为2g(以虎杖水提药渣提取物计),共洗脱5个柱体积,收集洗脱液,目测色带分离情况并tlc检测含虎杖苷和大黄素的组分,合并得到虎杖苷粗品和大黄素粗品。
[0069]
表2
[0070][0071]
径高比过高,虎杖苷和大黄素组分不能完全分离,随着径高比的减少,虎杖苷和大黄素的分离度增加,但是柱长过长分离时间增加,径高比以1:50~200为宜。甲醇系统、氯仿
‑
甲醇1:1系统、丙酮系统和75vol%甲醇水溶液系统都可以完全分离虎杖苷和大黄素组分,分离度以甲醇系统最好。氯仿
‑
甲醇1:1系统,需要用到毒性有机溶剂氯仿。丙酮系统sephadex lh
‑
20溶胀系数低,柱体积减小,载样量降低。75vol%甲醇水溶液系统样品的溶解性不好,上样体积增大,分离度降低。
[0072]
对比例2
[0073]
取5g虎杖水提药渣提取物上硅胶柱(硅胶填料100g,径高比1:10),依次以氯仿
‑
甲醇10:1、氯仿
‑
甲醇5:1、氯仿
‑
甲醇2:1、氯仿
‑
甲醇1:1梯度洗脱,每个梯度洗脱4个柱体积,收集洗脱液,tlc检测含虎杖苷和大黄素的组分,合并得虎杖苷粗品和大黄素粗品。结果显示,虎杖苷和大黄素组分有交叉,未能完全分离。
[0074]
虎杖苷和大黄素在硅胶填料上拖尾较严重,且硅胶柱需要用到氯仿等毒性有机溶剂。
[0075]
对比例3
[0076]
取5g虎杖水提药渣提取物上c
18
色谱柱(c
18
填料100g,径高比1:10),依次以甲醇
‑
水1:9、甲醇
‑
水2:8、甲醇
‑
水3:7、甲醇
‑
水4:6、甲醇
‑
水5:5、甲醇
‑
水6:4、甲醇
‑
水7:3、甲醇
‑
水8:2和甲醇梯度洗脱,每个梯度洗脱3个柱体积,收集洗脱液,tlc检测含虎杖苷和大黄素的组分,合并得虎杖苷粗品和大黄素粗品。虎杖苷和大黄素组分可完全分离。分离时间:36h以上。
[0077]
虽然采用c
18
柱可以将虎杖苷和大黄素组分完全分离,但需要梯度洗脱,而且需洗脱二十余个柱体积,操作较繁琐,操作过程中需回收大量含水的溶剂,能耗较大,且完成实验周期长。
[0078]
实施例9
[0079]
将虎杖苷对照品过滤后上agilent 1260高效液相色谱仪,紫外检测器在线监测,确定虎杖苷保留时间和峰形。将实施例3得到的虎杖苷粗品过滤后上agilent1260高效液相
色谱仪,根据虎杖苷对照品的保留时间和峰形,针对性收集虎杖苷色谱峰段制备溶剂,回收溶剂,得虎杖苷100mg。
[0080]
虎杖苷对照品和虎杖苷粗品的色谱条件为:色谱柱:c
18
色谱柱(9.4mm
×
250mm,5μm);流动相:甲醇a与水b;梯度洗脱程序:0~30min,10vol%~20vol%a;流速:2ml/min。
[0081]
实施例10
[0082]
将虎杖苷对照品过滤后上agilent 1260高效液相色谱仪,紫外检测器在线监测,确定虎杖苷保留时间和峰形。将实施例3得到的虎杖苷粗品过滤后上agilent1260高效液相色谱仪,根据虎杖苷对照品的保留时间和峰形,针对性收集虎杖苷色谱峰段制备溶剂,回收溶剂,得虎杖苷0.5g。
[0083]
虎杖苷对照品和虎杖苷粗品的色谱条件为:色谱柱:c
18
色谱柱(30mm
×
250mm,10μm);流动相:乙腈a与水b;等度洗脱程序:10vol%a;流速:10ml/min。
[0084]
实施例11
[0085]
将虎杖苷对照品过滤后上agilent 1260高效液相色谱仪,紫外检测器在线监测,确定虎杖苷保留时间和峰形。将实施例3得到的虎杖苷粗品过滤后上agilent1260高效液相色谱仪,根据虎杖苷对照品的保留时间和峰形,针对性收集虎杖苷色谱峰段制备溶剂,回收溶剂,得虎杖苷1.5g。
[0086]
虎杖苷对照品和虎杖苷粗品的色谱条件为:色谱柱:c
18
色谱柱(50mm
×
250mm,10μm);流动相:甲醇a与水b;等度洗脱程序:15vol%a;流速:30ml/min。
[0087]
实施例12
[0088]
将大黄素对照品过滤后上agilent 1260高效液相色谱仪,紫外检测器在线监测,确定大黄素保留时间和峰形。将实施例3得到的大黄素粗品过滤后上agilent1260高效液相色谱仪,根据大黄素对照品的保留时间和峰形,针对性收集大黄素谱峰段制备溶剂,回收溶剂,得大黄素400mg。
[0089]
大黄素对照品和大黄素粗品的色谱条件为:色谱柱:c
18
色谱柱(9.4mm
×
250mm,5μm);流动相:甲醇a与水b;梯度洗脱程序:0~30min,70vol%~90vol%a;流速:2ml/min。
[0090]
实施例13
[0091]
将大黄素对照品过滤后上agilent 1260高效液相色谱仪,紫外检测器在线监测,确定大黄素保留时间和峰形。将实施例3得到的大黄素粗品过滤后上agilent1260高效液相色谱仪,根据大黄素对照品的保留时间和峰形,针对性收集大黄素谱峰段制备溶剂,回收溶剂,得大黄素2.0g。
[0092]
大黄素对照品和大黄素粗品的色谱条件为:色谱柱:c
18
色谱柱(30mm
×
250mm,10μm);流动相:乙腈a与水b;等度洗脱程序:60vol%a;流速:10ml/min。
[0093]
实施例14
[0094]
将大黄素对照品过滤后上agilent 1260高效液相色谱仪,紫外检测器在线监测,确定大黄素保留时间和峰形。将实施例3得到的大黄素粗品过滤后上agilent1260高效液相色谱仪,根据大黄素对照品的保留时间和峰形,针对性收集大黄素谱峰段制备溶剂,回收溶剂,得大黄素7.0g。
[0095]
大黄素对照品和大黄素粗品的色谱条件为:色谱柱:c
18
色谱柱(50mm
×
250mm,10μm);流动相:甲醇a与水b;等度洗脱程序:80vol%a;流速:30ml/min。
[0096]
实验例
[0097]
1.结构确认
[0098]
将实施例9~14得到的虎杖苷和大黄素进行核磁共振实验,所得谱图数据如下:
[0099]
大黄素:橙色结晶(甲醇)。
13
c
‑
nmr(125mhz,dmso
‑
d6)δ:189.8(c
‑
9),181.4(c
‑
10),165.7(c
‑
3),164.6(c
‑
1),161.5(c
‑
8),148.4(c
‑
6),135.1(c
‑
4a),132.9(c
‑
10a),124.2(c
‑
7),120.6(c
‑
5),113.5(c
‑
8a),108.9(c
‑
9a),108.8(c
‑
4),107.9(c
‑
2),21.5(3
‑
ch3)。以上数据与文献报道一致(王乐,范书生,王秀环,等.女儿茶化学成分研究.中草药,2019,50(21):5217
‑
5222.)。
[0100]
虎杖苷:白色针状结晶(甲醇)。
13
c
‑
nmr(125mhz,dmso
‑
d6)δ:60.8(c
‑6″
),69.8(c
‑4″
),73.3(c
‑2″
),76.8(c
‑5″
),77.1(c
‑3″
),100.8(c
‑1″
),102.8(c
‑
4),104.8(c
‑
2),107.3(c
‑
6),115.7(c
‑3′5′
),125.2(c
‑
α),128.0(c
‑2′6′
),128.2(c
‑
β),128.6(c
‑1′
),139.5(c
‑
1),157.4(c
‑4′
),158.4(c
‑
5),159.0(c
‑
3)。以上数据与文献报道一致(杨华良,庾石山,裴月湖.短萼仪花叶化学成分研究.中国中药杂志,2008,33(22):2633
‑
2635.)。
[0101]
2.纯度检测
[0102]
检测实施例3中的虎杖苷粗品和大黄素粗品的纯度,实施例9~14中虎杖苷和大黄素的纯度。
[0103]
供试品溶液的制备:精密称定样品,加甲醇制成每1ml含100μg样品的储备液,稀释得供试品溶液。
[0104]
色谱条件:色谱柱:acquity beh c18色谱柱(2.1mm
×
100mm,1.7μm);流动相:乙腈a与0.2vol%甲酸溶液b;梯度洗脱:0~7min,12~20vol%a;7~10min,20~28vol%a;10~12min 28vol%a;12~15min 28~30vol%a;15~16min 30~80vol%a;16~18min 80vol%a);流速:0.4ml/min;柱温为40℃;检测波长为290nm。
[0105]
检测方法:精密吸取供试样品溶液,注入acquity uplc液相色谱仪,得到液相色谱图,以峰面积归一法测定样品的纯度,所得结果如表3所示。
[0106]
表3
[0107]
序号纯度/wt%实施例3
‑
大黄素粗品80.2实施例3
‑
虎杖苷粗品81.5实施例999.6实施例1099.5实施例1199.3实施例1299.7实施例1399.5实施例1499.5
[0108]
本发明并不限于上述实施方式,在不背离本发明的实质内容的情况下,本领域技术人员可以想到的任何变形、改进、替换均落入本发明的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

