1.本技术涉及寡核苷酸领域,具体而言,涉及一种亚磷酰胺化的核苷衍生物及其合成方法、应用和试剂盒。
背景技术:
2.迄今为止,对于癌症、病毒性感染等疾病,临床上仍缺少理想的特效药物。可喜的是,随着人类及重要模式生物基因组测序的完成,以及功能基因组学及蛋白质组学研究的深入,与疾病相关的分子靶标不断被发现和认识,为基因治疗提供了前提。
3.在过去的几十年里,人工合成的寡核苷酸已经广泛应用于靶向基因治疗的研究。寡聚核苷酸主要包括反义寡核苷酸(antisense oligonucleotides,asodn)、小干扰rna(small interference rna,sirna)、转录因子诱饵(decoy)、核酶(ribozyme)、脱氧核酶(dnazyme)、反基因(antigene)、cpg寡核苷酸和核酸适配体(aptamer)等。其中,asodn和sirna是最常用的基因调控工具,获得广泛应用,并已被开发为基因治疗药物。
4.目前,寡核苷酸主要采用化学方法合成。当前的化学合成工艺存在初产品纯度低,难以纯化,大量制备成本十分高昂,极大限制了寡核苷酸药物的研究和应用。
技术实现要素:
5.本技术提供了一种亚磷酰胺化的核苷衍生物及其合成方法、应用和试剂盒。该合成方法方便实施,且实施成本低,利于大规模生产。
6.本技术是这样实现的:
7.在第一方面,本技术的示例提供了一种亚磷酰胺化的核苷衍生物的合成方法。其以腺嘌呤2
′‑
脱氧核苷为起始物,经过基团保护、脱保护以及修饰目标基团而实现。
8.合成方法包括:
9.步骤1:对起始物的3
′
和5
′
羟基保护,再对碱基上的n6位氨基进行保护,然后脱除3
′
和5
′
羟基的保护基团,获得第一中间体;
10.步骤2:依次对第一中间体的5
′
和3
′
羟基进行保护,获得第二中间体;
11.步骤3:脱除第二中间体的5
′
羟基的保护基团,并通过甲氧基异丙基对5
′
羟基进行保护,获得第三中间体;
12.步骤4:依次脱除第三中间体中的3
′
羟基的保护基团和碱基上的n6位氨基的保护基团,获得第四中间体;
13.步骤5:依次对第四中间体的碱基上的n6位氨基进行亚胺化保护,3
′
羟基进行亚磷酰胺化保护,获得亚磷酰胺化的核苷衍生物。
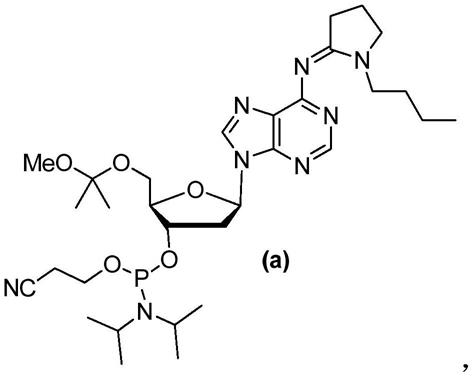
14.在第二方面,本技术示例提出了一种亚磷酰胺化的核苷衍生物,衍生物具有下述结构:
[0015][0016]
在第三方面,本技术示例提出了一种亚磷酰胺化的核苷衍生物在合成反义寡聚核苷酸中的应用。
[0017]
在第四方面,本技术示例提出了一种试剂盒,含有前述的亚磷酰胺化的核苷衍生物或其制成品。
[0018]
在以上实现过程中,本技术实施例提供的合成方法具有工艺过程简单易于实施的特点。并且,该合成方法各步骤中的产物纯度高、易于纯化,且实施成本低,利于大量生产。
[0019]
此外,本技术的示例合成方法还至少具有以下一些优点:
[0020]
1、本技术提供的制备方法初始原料为脱氧腺嘌呤核苷,来源广泛,经济易得。
[0021]
2、本技术提供的制备方法无需特殊设备,操作简单方便,适合大规模工业化生产。
[0022]
3、本技术提供的制备方法仅仅只需要结晶和硅胶纯化就能得到高纯度的核苷亚磷酰胺,具有良好的工业应用价值。
具体实施方式
[0023]
核苷亚磷酰胺是用于合成寡核苷酸的天然或合成核苷的衍生物。通常而言,当核苷或其类似物中含有至少一个羟基时,通过使用适当的基团保护策略就可以将核苷或其类似物转化成各自的亚磷酰胺。并且,后续可以以此亚磷酰胺化的核苷或其类似物为原料,通过反应而者并入合成的核酸中。
[0024]
亚磷酰胺的一个重要特征是它们在酸性催化剂存在下进行亚磷酰胺偶联反应的能力。这使得核苷亚磷酰胺成为在寡核苷酸合成中有用的中间体。因此,在寡核苷酸领域中,迫切需要适合大规模生产修饰性核苷亚磷酰胺的合成技术。
[0025]
有鉴于此,本技术发明人,在本技术中提出了一种亚磷酰胺化的核苷衍生物的合成方法。示例中,该衍生物是核苷亚磷酰胺;更具体而言,其是5'-o-2-mip(甲氧基丙基)保护的腺嘌呤脱氧核苷亚磷酰胺,且具有如下述式a的结构。
[0026]
其中的me表示甲基。
[0027]
根据上述可知,该衍生物以腺嘌呤脱氧核苷酸为主体,且分别对腺嘌呤的碱基上的胺进行保护,核糖的3'羟基进行了亚磷酰胺化,核糖的5'羟基进行了保护。
[0028]
因此,该衍生物可以作为制作其他核苷酸的中间体被使用。换言之,在本技术中,发明人提出了一种新的中间体,其可以被用于合成核苷酸。其中,碱基上的氨基保护和5'羟基保护都可以方便地在合成核苷酸的过程中予以脱除。而3'羟基的亚磷酰胺化则可以用以构成核苷酸中的磷酸二酯键。例如,一些二聚核苷酸中,两个核糖的3'碳和5'碳通过磷酸二酯键连接。因此,该化合物a可以作为合成反义寡聚核苷酸中的原料中间体。除此之外,其还可以被用于制作试剂盒。在该试剂盒中,化合物a可以是衍生物—即其制成品—形式存在。
[0029]
在化学合成中,尤其是核苷酸领域,可以通过采用各种中间体来缩短合成路径、简化合成方法。但是,这些方案中往往需要特殊的中间体,从而导致工艺成本非常高,或者说由于中间体的结构特殊性导致产物的收率很低、杂质含量较高。
[0030]
有鉴于这样的显示状况,在本技术示例中,以基础的腺嘌呤脱氧核苷为起始物通过巧妙地选择合成路径,以期望的优势获得了目标产物(前述a式化合物)。
[0031]
其中的,起始物具有下述式b所示的结构。
[0032]
其中的核糖单元为2'-脱氧核糖。
[0033]
换言之,本技术示例中的一个目标是通过起始物b合成衍生物a,如下所示。
[0034]
[0035]
因此,从合成路径上而言,可以对n6位上的氨基的亚胺化保护(第一处理)、3'羟基的亚磷酰胺化(第二处理)和5'羟基的保护(第三处理)通过在不同的步骤中进行保护和脱保护,从而实现独立地对上述三个基团进行分别修饰。即通过基团保护和脱保护的方式,就n6位上的氨基、3'羟基和5'羟基而言,将需要进行处理的基团独自“暴露”,而将暂时不需要处理的基团进行“掩蔽”(通过保护基团将其沉默)。如此,被“暴露”的基团能够进行需要的反应处理(即第一处理,或第二处理,或第三处理)。
[0036]
因此,基于上述的合成思路,例如,一些可能的选择如下。
[0037]
第一选择:先实施第一处理,然后,再实施第二处理,其次实施第三处理。
[0038]
第二选择:先实施第一处理,然后,再实施第三处理,其次实施第二处理。
[0039]
第三选择:先实施第二处理,然后,再实施第三处理,其次实施第一处理。
[0040]
第四选择:先实施第二处理,然后,再实施第一处理,其次实施第三处理。
[0041]
第五选择:先实施第三处理,然后,再实施第一处理,其次实施第二处理。
[0042]
第六选择:先实施第三处理,然后,再实施第二处理,其次实施第一处理。
[0043]
由于上述不同的合成路径顺序,会涉及到不同的基团保护和脱保护方式,同时也会对反应过程中使用的试剂和反应后的纯化过程等产生具体且实质性的影响。
[0044]
通过实验探索和研究,发明人以上述第五选择的方式实施合成方法。即,先实施第三处理,然后,再实施第一处理,其次实施第二处理。对应于此,对基团的掩蔽和暴露方式进行巧妙的选择,从而实现了以基础起始物(而非借助于的中间体)为原料,低成本和高效地合成了所设计的5'-o-2-mip(甲氧基丙基)保护的修饰性核苷亚磷酰胺—a式化合物。
[0045]
整体上而言,本技术示例中的合成方法可以通过下述描述被阐明。
[0046]
(1):以腺嘌呤脱氧核苷(b)作为原料,先将其3'位和5'位的羟基用硅烷化试剂进行保护,然后再将碱基(腺嘌呤)的n6位的氨基进行保护,接下来再脱除3位和5位的保护,得到中间体(c)。下述反应方程式中略去了,在3'位和5'位上保护基团的过程。
[0047][0048]
其中的r1表示氨基保护基,且可以选择为ac(乙酰基),bz(苯甲酰基),pac(苯氧乙酰基),fmoc(9-芴甲氧羰基)等保护基。
[0049]
其中,用于对核糖上的羟基进行保护的试剂例如可以选择为硅烷化试剂。示例性,硅烷化试剂包括但不限于tms-cl(三甲基氯硅烷),tbdms-cl(叔丁基二甲基氯硅烷),tbdps-cl(叔丁基二苯基氯硅烷),tips-cl2(三异丙基氯甲硅烷)或其组合(两种或以上的混合物)。其中,以tms-cl为佳。这些催化剂的用量不宜太多,也不宜太少,以能够将起始物转化为羟基保护形式的产物为限。例如,示例中,起始物与上述硅烷化试剂的摩尔用量之比可以控制在1:2.5至1:4.0;例如摩尔比可以为1:2.6,1:2.8,1:3,1:3.5或1:3.8等;当采用的是复合的保护试剂时,上述摩尔比是以硅烷化试剂中硅的摩尔量计。
[0050]
并且在进行保护时,上述硅烷化试剂是在有机溶剂体系中进行的;进一步地,是在催化剂的有机溶剂体系中进行的。例如,选择吡啶为催化剂,并提供液态体系。或者,以三乙胺为催化剂,并且是二氯甲烷或已腈的溶液体系。这些有机溶剂体系的用量可以限制为起始物与有机溶剂的液料比(v:m)为4:1至10:1。
[0051]
在进行上述的两个碳位的羟基保护(羟基掩蔽或羟基沉默)之后,对腺嘌呤上的n6位氨基保护(处于暴露状态)的试剂可以选择位accl,bzcl,paccl或fmoccl等试剂。这些氨基保护试剂的用量可以选择为相对于起始物适当过量;即氨基保护基团的摩尔量大于氨基的摩尔量。例如,起始物与r1cl的摩尔比为1:1.05-1:1.50。
[0052]
随后,为了释放羟基以备后续反应,在氨基保护之后对羟基保护基团进行脱除。示例中,对应于上述的硅烷化试剂,使用的是硅脱保护试剂。例如,tbaf(四丁基氟化铵),tea
·
3hf(三乙胺三氢氟酸盐),hf
·
pyridine(氟化氢-吡啶络合物,或氟化氢吡啶溶液)。而硅脱保护试剂的用量可以是起始物摩尔量的2倍或五倍。
[0053]
通过上述反应之后可以获得式c所示的化合物。并且,为了简化操作两个碳位的羟基的保护通过一锅法实现,从而可以减少分离和纯化的操作,也减少物料的使用。然后再向其中加入氨基保护试剂进行反应,随着再向其中加入脱保护试剂经过简单的分离纯化获得c。其中的分离纯化操作例如是通过过滤,将反应体系中的不溶的固体滤除,然后对滤液进行浓缩获得粗品。随后,通过重结晶的方式获得c。其中的重结晶例如采用乙酸乙酯/正庚烷的混溶试剂,其用量以维持在起始物的质量的5倍至10倍为宜。
[0054]
(2):将中间体c的5'-oh进行保护,获得中间体(d):
[0055][0056]
其中,r2为羟基保护基,可以为dmtr(二对甲氧基三苯甲基),mmtr(对甲氧基苯基二苯甲基),bz(苯甲酰基)等;相应地,保护试剂可以是r2cl,示例性地包括dmtrcl(二对甲氧基三苯甲基氯),mmtrcl(对甲氧基苯基二苯甲基氯),bzcl(苯甲酰氯)等。化合物c与r2cl的比例为1;1.0-1:1.5。催化剂则可以采用吡啶、三乙胺等,而作为溶剂则可以是dmf(二甲基甲酰胺)、二氯甲烷。
[0057]
(3)将中间体d和硅烷化试剂进行反应,将3'-oh进行硅烷化保护,得到中间体e。
[0058]
[0059]
r3表示硅保护基,可以为tms(三甲基硅基),tbdms(叔丁基二甲基硅基),tes(三乙基硅基),tbdps(叔丁基二苯基硅基)等。对应所使用的保护试剂为r3cl,示例性地包括tmscl(三甲基氯硅烷),tbdmscl(叔丁基二甲基氯硅烷),tescl(三乙基氯硅烷),tbdpscl(叔丁基二苯基氯硅烷)等。化合物d与r3cl按照摩尔比1:1.0-1:1.5的比例用料。反应时,采用的溶剂可以是吡啶,吡啶/dmf,吡啶/dmso,吡啶/二氯甲烷,二氯甲烷/三乙胺等。并且,溶剂的与为化合物d的液料比为5至15。
[0060]
(4):将中间体e用酸脱除5
′‑
oh的保护基,得到中间体f。
[0061][0062]
该步骤对5
′‑
oh的保护基采用tfa/meoh(三氟乙酸和甲醇溶液),或dca/c
12h25
sh(二氯乙酸和叔十二烷基硫醇溶液)等进行脱除。其中的脱保护试剂的用量(以三氟乙酸或二氯乙酸为准)为化合物d的摩尔量的2倍或5倍。反应完成之后,通过过滤、浓缩获得粗品,再通过重结晶获得化合物f。其中的粗品可以用乙酸乙酯/二氯甲烷混合溶液作为溶剂进行结晶;更佳地,按照重量体积比,结晶溶剂用量为化合物d重量的2-6倍。
[0063]
(5)中间体f在酸催化下和二甲氧基丙烷反应,将5
′‑
oh用mip保护基保护起来,得到中间体g。
[0064][0065]
其中的mip是保护基团,且为甲氧基异丙基。示例中,用酸催化二甲氧基丙烷与5
′‑
oh进行反应而实现保护。其中的酸催化剂例如选自:硫酸、三氟甲磺酸、三氟甲磺酸银、三氟甲磺酸钪等。部分示例中,酸催化剂和化合物f按照摩尔比1:100至10:100。
[0066]
(6)中间体(g)脱除3-oh的硅保护基,得到中间体(h)。
[0067][0068]
脱除3'-oh的硅保护基所使用的脱保护试剂,例如选自tbaf,tea
·
3hf,hf
·
pyridine等。并且其中的化合物g与脱保护试剂按照摩尔比1:1.5-1:8.0的配比。
[0069]
(7)中间体(h)脱除n6位氨基的bz保护,得到中间体(i)。
[0070][0071]
采用脱保护试剂脱除中间体(h)的n6位氨基的保护基。脱保护试剂例如选自氨水,tea(三乙胺),naoh,k2co3,甲基哌嗪等。部分示例中,化合物h与脱保护试剂摩尔比为1:5至1:30。
[0072]
(8)中间体(i)的n6位氨基和缩醛进行反应得到中间体(j)。
[0073][0074]
中间体(i)的n6位氨基用缩醛保护形成亚胺;中间体(i)与缩醛的摩尔比为1:2-1:10。其中的缩醛包括但不限于1-丁基-2,2-二甲氧基吡咯烷,cas no.:74255-10-0;1-甲基-2,2-二甲氧基吡咯烷,cas no.:39650-82-3;1,1-二甲氧基-n,n-二甲基乙胺,cas no.:18871-66-4;n,n-二甲基甲酰胺二甲基缩醛,cas no.:4637-24-5等。
[0075]
(9)中间体(j)进行亚磷酰胺化得到最终的修饰性核苷亚磷酰胺(a)。
[0076][0077]
中间体(j)的3'-oh与磷试剂反应生成亚磷酰胺。其中的磷试剂可以选用双二异丙基氨基氯化磷或双(二异丙基氨基)(2-氰基乙氧基)膦;一些示例中,中间体(j)与磷试剂的摩尔比为1:0.9-1:2.0。
[0078]
本技术的上述示例方案中,考虑到亚胺结构不稳定,且遇酸碱都有降解的风险。因此,如果对上述分子b先上亚胺保护,那么后续在5
′‑
位羟基进行mip保护时,需要用tsoh催化,而此时亚胺会被去保护。进一步地,考虑到亚磷酰胺这一结构也是酸碱都不稳定,且纯
化方式有限,因此,选择路线设计中在最后一步才会进行亚磷酰胺化反应,有助于降低纯化难度,也避免其收率太低。此外,在分子i的基础上先进行亚胺保护,可以确保化合物j的纯度足够高,然后再进一步进行亚磷酰胺化,大大降低了进行纯化操作以获得化合物a的难度,从而能够确保能够拿到磷谱和hplc都大于98%以上的产品。
[0079]
应当指出的是,在前文的描述中,所有以数值范围形式界定的特征—如数值、数量、含量与浓度仅是为了简洁及方便。据此,数值范围的描述应视为已涵盖且具体公开所有可能的次级范围及范围内的个别数值(包括整数与分数)。
[0080]
此外,在前文所描述的各步骤中的各种试剂,在不同的实施例中,可以适应性地调整各个步骤中的试剂。换言之,上述特征,或示例所提到的特征可以任意组合。本案说明书所揭示的所有特征可与任何组合物形式并用,只要这些特征的组合不存在矛盾,所有可能的组合都应当认为是本说明书记载的范围。说明书中所揭示的各个特征,可以任何可提供相同、均等或相似目的的替代性特征取代。因此除有特别说明,所揭示的特征仅为均等或相似特征的一般性例子。
[0081]
下面将结合实施例对本技术的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本技术,而不应视为限制本技术的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0082]
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。
[0083]
实施例1
[0084]
中间体c的合成:
[0085][0086]
操作步骤:
[0087]
将化合物b(190g,0.756mol,1.0eq.)溶于吡啶(1.9l)中,加入tmscl(329g,3.024mol,4.00eq.),25
±
5℃反应2个小时后。
[0088]
然后加入bz2o(过氧化二苯甲酰,342g,1.512mol,2.0eq.),反应液升温至60
±
5℃搅拌8小时后,加水(400ml)搅拌1小时。
[0089]
随后,反应液直接浓缩干,加入ea(乙酸乙酯,1.9l)复溶,每次用水(1l)洗涤,共洗涤两次。洗涤后进行分液,并将分出的有机相用无水硫酸钠干燥,经过35
±
5℃浓缩干,得到粗品。粗品用etoh/ea(乙醇:乙酸乙酯;1.8l/0.2l)悬浮搅拌30分钟,过滤,真空干燥后得到200g化合物c,hplc纯度为98.5%,摩尔收率:74%。
[0090]
化合物c的确认:
[0091]
h nmr(500mhz,dmso-d6)d 11.2(s,br,1h),8.75(s,1h),8.65(s,1h),8.0(d,1h),
7.5
–
7.6(m,4h),6.4(dd,1h),5.6(s,br,1h),4.7(s,br,1h),4.4(m,1h),4.0(m,1h),3.6
–
3.7(m,2h),2.8(m,1h),2.3(m,1h).
[0092]
hrms(esi)calcd for c17h18n5o4[m h] :356.1314;found:356.1316。
[0093]
中间体d的合成:
[0094][0095]
操作步骤:
[0096]
将化合物(c)(200g,0.563mol,1.00eq.)溶于吡啶(1l)和乙腈(1l)的混合溶液中,然后于45
±
5℃减压浓缩至不滴,得到油状物。
[0097]
然后,向油状物中加入dmso(二甲基亚砜,1l)和吡啶(0.2l)溶解,然后将反应液降温至10
±
2℃。向反应液中分三批加入dmtrcl(190g,0.563mol,1.00eq.),控制反应内温不超过12℃,滴加完后,反应12小时。
[0098]
反应结束后,取样检测,要求原料(化合物c)<3wt%则反应合格。向反应液中加入甲醇(40ml),搅拌15-20分钟。然后再均分三次加入碳酸氢钠(100g)固体,每次间隔10分钟左右,加完继续搅拌30分钟。随后,将反应混合物于45
±
5℃减压浓缩去除吡啶,加入dcm(800ml)和水(800ml)萃取分液。分离出的有机相分别用5%的醋酸(800ml
×
2)和5%的碳酸氢钠溶液(800ml)洗涤,然后有机相用无水硫酸钠干燥除水。干燥后的有机相浓缩干,得到泡沫状固体。将泡沫状固体加乙腈(1l)和dcm(0.1l)结晶,过滤,真空干燥后得到300g产品(d),hplc纯度为99.0%,摩尔收率:80%。
[0099]
化合物d的确认:
[0100]1h nmr(500mhz,cdcl3)δ10.88(s,1h),8.34(s,1h),8.20(s,1h),7.96
–
7.89(m,2h),7.70
–
7.49(m,3h),7.36
–
7.32(m,2h),7.31
–
7.29(m,2h),7.27
–
7.24(m,5h),6.92
–
6.79(m,4h),6.65(t,j=15.0hz,1h),4.59(td,j=12.4,2.9hz,1h),4.49-4.39(m,1h),3.96(dd,j=24.7,12.5hz,1h),3.86-3.83(m,1h),3.78(s,6h),3.56(s,1h),3.54(dd,j=24.7,12.5hz,1h),),2.54-2.48(m,1h)。
[0101]
hrms(esi)calcd for c38h35n5o6[m h] :658.2621;found:658.2624.
[0102]
中间体f的合成:
[0103][0104]
操作步骤:
[0105]
中间体e的制备:
[0106]
将中间体d(198.0g,1.0eq.)加入乙腈(1l
×
3)浓缩带干。浓缩干后,加dcm(2.9l)
悬浮,温度降至0-10℃。往上述反应液中先加入咪唑(61.49g,3.0eq.),再加tbdmscl(113.43g,2.5eq.),加完后于25℃反应。反应12小时后,取样tlc监控,原料无剩余。
[0107]
中间体f的制备:
[0108]
向反应液中加入甲醇(198g)淬灭反应,搅拌30分钟后直接进入下一步。将反应液降温至0-5℃,滴加tfa(103.14g,3.0eq.),加完后于0-5℃反应。反应1.5小时后,取样tlc监控,原料无剩余。
[0109]
分离纯化:将反应液直接倒入5%nahco3水溶液(2l)中搅拌30分钟,分出有机相,用5%nahco3水溶液(2l)洗一次,用10%氯化钠水溶液(2l)洗一次,分出有机相,用无水硫酸钠干燥,过滤后浓缩干得到粗品。随后,往粗品中加入ea/dcm(350ml/100ml)的混合溶剂溶解,再缓慢加入正庚烷(2.12l),加完后于10-15℃搅拌结晶,8小时后过滤,得到固体。对该固体进行高真空干燥,得到130g中间体f,uplc纯度:97.62%。摩尔收率:92.2%。
[0110]
中间体f的确认:
[0111]1h nmr(500mhz,cdcl3)δ10.95(s,1h),8.34(s,1h),8.20(s,1h),7.95
–
7.88(m,2h),7.69
–
7.47(m,3h),7.36
–
7.33(m,2h),7.32
–
7.30(m,2h),7.27
–
7.24(m,5h),6.92
–
6.79(m,4h),6.65(t,j=15.0hz,1h),4.59(td,j=12.4,2.9hz,1h),3.96(dd,j=24.7,12.5hz,1h),3.86-3.83(m,1h),3.78(s,6h),3.58(dd,j=24.7,12.5hz,1h),2.59-2.50(m,1h),1.96-1.83(m,1h),0.98(s,9h),0.21(s,6h).hrms(esi)calcd for c23h31n5o4si[m h] :470.2179;found:470.2183.
[0112]
中间体g的合成:
[0113][0114]
操作步骤:
[0115]
将中间体f(130g,1.0eq.)加入thf(1300ml,10v)溶解,加二甲氧基丙烷(650ml,5v),再加入ts-oh(5.27g,0.1eq.),加完后于25℃反应。反应2.5小时后,向反应液中加入tea(100ml)淬灭反应,搅拌30分钟。
[0116]
分离纯化:将反应液直接浓缩干得到粗品,粗品用硅胶层析,硅胶预先用2%的三乙胺碱化,洗脱梯度乙酸乙酯/正庚烷=1/4-1/2,纯产品收集后,于35
±
5℃高真空减压浓缩至不滴得到白色固体,高真空干燥后得到90g中间体f,uplc:98.81%,w.y.:69.2%m.y:60.0%。
[0117]1h nmr(500mhz,cdcl3)δ10.87(s,1h),8.33(s,1h),8.19(s,1h),7.95
–
7.88(m,2h),7.67
–
7.47(m,3h),6.64(t,j=15.0hz,1h),4.59(td,j=14.7,0.8hz,1h),3.94(dd,j=24.7,14.7hz,1h),3.84(td,j=4.6,0.9hz,1h),3.76(dd,j=24.7,14.8hz,1h),3.39(s,3h),2.42-2.22(m,2h),1.21(s,6h),0.98(s,9h),0.21(s,6h)。
[0118]
hrms(esi)calcd for c27h39n5o5si[m h] :542.2754;found:542.2751.
[0119]
中间体h的合成:
[0120][0121]
操作步骤:
[0122]
中间体g(90.0g,1.0eq.),加thf(900ml)溶解,再加入tbaf(86.88g,2.0eq.),加完后于25℃反应。反应1.5h后,tlc监控,原料无剩余。
[0123]
分离纯化:反应液于35
±
5℃浓缩至无冷凝液滴下,加dcm(900ml)溶解后水洗1次,水相用f001(700ml)反萃一次,合并有机相,有机相干燥过滤浓缩干后得到65g中间体h。
[0124]
1h nmr(500mhz,cdcl3)δ10.52(s,1h),8.35(s,1h),8.21(s,1h),7.96-7.92(m,2h),7.65
–
7.50(m,3h),7.07(t,j=8.4hz,1h),4.64-4.56(m,1h),4.13-4.02(m,2h),3.56(dd,j=24.9,10.6hz,1h),3.48(s,1h),3.40(s,3h),2.82-2.72(m,1h),2.03-1.93(m,1h),1.21(s,6h)。
[0125]
hrms(esi)calcd for c21h21n5o5[m h] :428.1889;found:428.1892.
[0126]
中间体i的合成:
[0127][0128]
操作步骤:
[0129]
中间体h(65g,1.0eq.),加甲醇(450ml)溶解,再加入氨水(900ml),加完后于50
±
5℃反应。反应6h后,取样tlc监控,原料无剩余。
[0130]
分离纯化:将反应液于35
±
5℃浓缩干得到粗品,粗品再用甲醇(250ml)带干两次,加乙腈(180ml)悬浮搅拌30分钟后,过滤,滤饼用甲醇洗涤,滤饼真空干燥后得到45g中间体i。
[0131]1h nmr(500mhz,cdcl3)δ8.58(s,1h),8.35(s,1h),7.35(s,2h),6.73(t,j=15hz,1h),4.60(td,j=14.5,1.7hz,1h),4.31(td,j=5.5,1.6hz,1h),4.17(dd,j=24.8,14.6hz,1h),3.56(s,1h),3.46(dd,j=24.7,14.5hz,1h),3.40(s,3h),2.51-2.42(m,1h),2.30-2.20(m,1h),1.21(s,6h)。
[0132]
hrms(esi)calcd for c14h21n5o4[m h] :324.1627;found:324.1625.
[0133]
中间体j的合成:
[0134][0135]
操作步骤:
[0136]
中间体i(45g,1.0eq.),加乙腈(450ml)带水三次,再加入甲醇(450ml)搅拌悬浮,加入1-丁基-2,2-二甲氧基吡咯烷,(cas no.:74255-10-0,59.94g,2.3eq.),加完后于25
±
5℃搅拌反应。反应2小时后,取样tlc监控,原料无剩余。
[0137]
分离纯化:将反应液直接35
±
5℃浓缩干得到粗品。粗品用硅胶层析,硅胶预先用2%的三乙胺碱化,洗脱梯度dcm/meoh=100/0-100/3,纯产品收集后,于35
±
5℃高真空减压浓缩至不滴,得到51g无色油状物。
[0138]1h nmr(500mhz,cdcl3)δ8.64(s,1h),8.32(s,1h),6.68(t,j=15hz,1h),4.4.63-4.54(m,1h),4.11-3.94(m,2h),3.62-3.55(m,3h),3.52(s,1h),3.46
–
3.25(m,5h),2.97(t,j=15hz,2h),2.77-2.67(m,1h),1.99-1.82(m,3h),1.55-1.43(m,2h),1.36-1.22(m,2h),1.15(s,6h),0.90(t,j=15hz,3h).
[0139]
hrms(esi)calcd for c22h34n6o4[m h] :447.2675;found:447.2677.
[0140]
目标产物a的合成:
[0141][0142]
操作步骤:
[0143]
化合物j(46.0g,1.0eq.),加入乙腈(460ml
×
3)带水,加dcm(460ml)溶解,加入双(二异丙基氨基)(2-氰基乙氧基)膦(34.13g,1.1eq)和四氮唑(5.77g,0.8eq.),加完后于25
±
5℃反应。反应1小时后,tlc监控,原料反应完。
[0144]
分离纯化:
[0145]
用5%的碳酸氢钠溶液(460ml
×
2)洗涤,10%的氯化钠溶液(460ml)洗涤,分出有机相,无水硫酸钠干燥,有机相于35
±
5℃减压浓缩得到粗品。
[0146]
粗品用硅胶柱层析,硅胶预先用2%的三乙胺碱化,洗脱梯度ea/n-heptane=1/1-8/1,纯产品收集后,于35
±
5℃高真空减压浓缩至不滴得45g无色油状物,产品为两种非对映异构体的混合物。
[0147]1h nmr(500mhz,cdcl3)δ8.45(s,1h),8.14(s,1h),6.45-6.42(m,1h),4.69-4.63(m,1h),4.23-4.17(m,1h),3.81-3.77(m,1h),3.73-3.67(m,1h),3.66
–
3.46(m,6h),3.40
(t,j=5hz,1h),3.05(s,3h),2.92-2.80(m,2h),2.74-2.67(m,1h),2.59-2.45(m,3h),1.97-1.90(m,3h),1.31-1.27(m,8h),1.13-1.11(m,12h),0.85(t,j=10hz,1h).
[0148]
hrms(esi)calcd for c
31h51
n8o5p[m h] :646.3720;found:646.3724.
[0149]
以上所述仅为本技术的优选实施例而已,并不用于限制本技术,对于本领域的技术人员来说,本技术可以有各种更改和变化。凡在本技术的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。