1.本技术的实施例涉及一种5,5-二甲基异噁唑烷-3-硫酮的制备方法,属于 医药或农药制备领域。
背景技术:
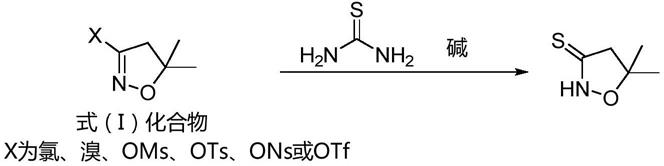
2.5,5-二甲基异噁唑烷-3-硫酮,是制备农化成分和药物成分的重要中间体, 目前,该化合物的合成工艺报道的不多,仅公开了以下工艺路线:
[0003][0004]
上述工艺路线以5,5-二甲基异恶唑烷-3-酮与有剧毒的五硫化二磷反应, 得到目标产物,该方法产生大量的磷、硫渣,三废多,难处理,反应恶心难 闻,不适于大规模生产。
技术实现要素:
[0005]
本技术实施例提供了一种5,5-二甲基异噁唑烷-3-硫酮的制备方法,该方 法简洁、高效,无需引入催化剂,反应条件温和、反应速度快、反应过程粗 放,对设备要求低,产物纯度高、收率高、产物易分离,具有很好的工业化 应用前景。
[0006]
本技术至少一个实施例提供了一种5,5-二甲基异噁唑烷-3-硫酮的制备 方法,所述方法包括:在溶剂存在下,将式(i)所示的化合物与硫脲和碱 接触以发生反应,
[0007][0008]
其中,式(i)中,x为氯、溴、甲基磺酰氧基、对甲苯磺酰氧基、三 氟甲磺酰氧基和对硝基苯磺酰氧基中的至少一种。
[0009]
根据一种优选的实施方式,式(i)所示的化合物、硫脲和碱的摩尔比 为1:(1-1.5):(2-3),进一步优选地,式(i)所示的化合物、硫脲和 碱的摩尔比为1:(1-1.2):(2-2.5)。
[0010]
根据一种优选的实施方式,反应的条件包括:温度为60-100℃,进一步 优选为60-80℃。
[0011]
根据一种优选的实施方式,所述溶剂为乙醇、正丁醇、甲醇、异丙醇、 叔丁醇和乙腈中的至少一种。
[0012]
根据一种优选的实施方式,式(i)所示的化合物与所述溶剂的质量比 为1:(1.5-10),进一步优选为1:(2-8)。
[0013]
根据一种优选的实施方式,所述碱为氢氧化钠、氢氧化钾和氢氧化锂中 的至少一种。
[0014]
根据一种优选的实施方式,所述反应中无催化剂存在。
[0015]
本技术实施例提供的5,5-二甲基异噁唑烷-3-硫酮的制备方法,该方法简 洁、高
效,无需引入催化剂,反应条件温和、反应速度快、反应过程粗放, 对设备要求低,产物纯度高、收率高、产物易分离,具有很好的工业化应用 前景。具体地,具有如下优点:
[0016]
(1)整个合成过程简洁、高效,反应条件温和,不需要特殊催化剂, 不需要过高的压力,对设备要求低,降低了反应的设备成本。
[0017]
(2)反应速度快,反应选择性高,产品纯度高。
[0018]
(3)合成过程和后处理过程简单粗放,反应得到的产物和废盐容易分 离,方便处理,反应的有机溶剂可以回用,适合工业化大生产。
[0019]
(4)代替了使用五氧化二磷的方法,反应条件温和,经济和环保价值 高。
具体实施方式
[0020]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发 明实施例,对本发明实施例的技术方案进行清楚、完整地描述。显然,所描 述的实施例是本发明的一部分实施例,而不是全部的实施例。基于所描述的 本发明的实施例,本领域普通技术人员在无需创造性劳动的前提下所获得的 所有其他实施例,都属于本发明保护的范围。
[0021]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这 些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各 个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点 值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视 为在本文中具体公开。
[0022]
本技术的至少一种实施例提供了一种5,5-二甲基异噁唑烷-3-硫酮的制 备方法,所述方法包括:在溶剂存在下,将式(i)所示的化合物与硫脲和 碱接触以发生反应,
[0023][0024]
其中,式(i)中,x为氯、溴、甲基磺酰氧基、对甲苯磺酰氧基、三 氟甲磺酰氧基和对硝基苯磺酰氧基中的至少一种。
[0025]
其中,本技术的反应式如下所示:
[0026][0027]
其中,本技术中,一种优选的实施方式中,5,5-二甲基异噁唑烷-3-硫酮 由式(i)所示的化合物、硫脲、碱通过一锅法反应得到。
[0028]
本技术中,上述式(i)中,x为离去基团,例如氯、溴、甲基磺酰氧 基(oms/mso)、对甲苯磺酰氧基(ots/tso)、三氟甲磺酰氧基(otf/tfo) 和对硝基苯磺酰氧基(ons)中的至少一种,其中,甲基磺酰氧基、对甲苯 磺酰氧基、三氟甲磺酰氧基、对硝基苯磺酰氧基的具体结构式如下:
[0029][0030]
本技术中,式(i)所示化合物均可通过现有方法制备得到,例如,x 为氯、溴时,可参照cn101389625a中实施例14所述的方法制备,x为甲 基磺酰氧基(oms)、对甲苯磺酰氧基(ots)、三氟甲磺酰氧基(otf) 和对硝基苯磺酰氧基(ons)时,可按照以下反应式制备得到:
[0031][0032]
此方法为本领域技术人员所公知,具体细节再此不再赘述,上述式(i) 所示化合物也均可通过商购获得。
[0033]
本技术中,为了减少杂质的生成量以及使碱更好的溶解,优选情况下, 式(i)所示的化合物、硫脲和碱的摩尔比为1:(1-1.5):(2-3),进一 步优选地,式(i)所示的化合物、硫脲和碱的摩尔比为1:(1-1.2):(2-2.5)。
[0034]
其中,对于式(i)所示的化合物、硫脲、溶剂和碱的加入顺序没有特 定的要求,只要将前述各物质进行混合即可,例如,可以先加入溶剂,再依 次加入硫脲、式(i)所示的化合物和碱。
[0035]
本技术中,为了兼顾反应原料的稳定性以及反应产物的高收率,优选情 况下,反应的条件包括:温度为60-100℃,进一步优选为60-80℃。为了使 反应更好的进行,优选在搅拌下进行反应。
[0036]
本技术中,反应条件温和、简单、高效,无须引入催化剂,反应原料在 反应溶剂以及合适的反应温度下即可发生反应。本技术所用的反应溶剂是为 原料的反应提供合适的反应环境,反应溶剂选择有机的、不与原料与产物反 应、能够很好的提供均相反应环境的溶剂即可,例如在有机反应中经常作为 溶剂使用的醇类溶剂、腈类溶剂等,包括但不限于乙醇、乙腈、甲醇、异丙 醇、正丁醇和叔丁醇等。反应溶剂能保证反应良好的进行即可,其使用量可 以根据反应容器和溶剂的利用率、反应效率、反应进行的困难度等情况进行 选择。优选情况下,式(i)所示的化合物与所述溶剂的质量比为1:(1.5-10), 进一步优选为1:(2-8)。
[0037]
本技术中,为了提高反应原料碱的溶解度以及加快反应速度,优选情况 下,所述碱为氢氧化钠、氢氧化钾和氢氧化锂中的至少一种。
[0038]
本技术中,可以通过hplc法来检测反应的进度,当检测到没有式(i) 所示的化合物时反应结束。将反应后的反应液,减压浓缩蒸除溶剂,冷却至 室温,除去无机盐,回收溶剂,得到5,5-二甲基异噁唑烷-3-硫酮。该后处理 过程简洁、粗放,产品和废盐容易分离。
[0039]
实施例
[0040]
如无特别说明,本技术中所用的材料均为本领域常用的各种相应材料, 均可商购获得。
[0041]
各实施例和对比例中,收率的计算公式为:收率=产物实际质量*纯度/ 产物理论质量。
[0042]
制备例1
[0043]
本制备例为5,5-二甲基-4,5-二氢-异噁唑-3基-甲磺酸酯的合成,即式(i) 化合物(x为oms)的合成。
[0044]
在反应四口瓶中加入二氯甲烷120g、5,5-二甲基-异噁唑烷-3酮11.5g和 三乙胺10.1g,搅拌冰水浴降温至0~5℃,滴加mscl 11.45g,滴毕,100ml 去离子水洗三次,无水硫酸钠干燥,减压浓缩得油状物18.5g,纯度为94.5%。
[0045]
制备例2
[0046]
本制备例为5,5-二甲基-4,5-二氢-异噁唑-3基-三氟甲基磺酸酯的合成, 即式(i)化合物(x为otf)的合成。
[0047]
在反应四口瓶中加入二氯甲烷120g、5,5-二甲基-异噁唑烷-3酮11.5g和 三乙胺10.1g,搅拌冰水浴降温至0~5℃,滴加三氟甲磺酸酐28.2g,滴毕,100ml去离子水洗三次,无水硫酸钠干燥,减压浓缩得油状物22.7g,纯度 为93.7%。
[0048]
制备例3
[0049]
本制备例为5,5-二甲基-4,5-二氢-异噁唑-3基-对甲基苯磺酸酯的合成, 即式(i)化合物(x为ots)的合成。
[0050]
在反应四口瓶中加入二氯甲烷g、5,5-二甲基-异噁唑烷-3酮11.5g和三 乙胺10.1g,搅拌冰水浴降温至0~5℃,滴加对甲苯磺酰氯19.1g,滴毕,100ml 去离子水洗三次,无水硫酸钠干燥,减压浓缩得油状物24.7g,纯度为94.1%。
[0051]
制备例4
[0052]
本制备例为5,5-二甲基-4,5-二氢-异噁唑-3基-对硝基苯磺酸酯的合成, 即式(i)化合物(x为ons)的合成。
[0053]
在反应四口瓶中加入二氯甲烷g、5,5-二甲基-异噁唑烷-3酮11.5g和三 乙胺10.1g,搅拌冰水浴降温至0~5℃,滴加对硝基苯磺酰氯22.2g,滴毕, 100ml去离子水洗三次,无水硫酸钠干燥,减压浓缩得油状物25.7g,纯度 为93.5%
[0054]
制备例5
[0055]
参照cn101389625a中实施例14所述的方法制备式(i)化合物(x为 氯)。
[0056]
制备例6
[0057]
参照cn101389625a中实施例14所述的方法制备式(i)化合物(x为 溴)。
[0058]
实施例1
[0059]
向反应四口瓶中加入甲醇50g、硫脲7.6g搅拌升温至66~68℃,加入式(i)所示化合物(x为氯)13.3g继续反应2hr,加入氢氧化钠8g,68℃反 应2hr,hplc(hplc测定仪器购自安捷伦公司,型号1260infinity ii,下 同)检测式(i)所示化合物完全转化,减压浓缩蒸除甲醇,冷却至室温, 加入40g水和36%浓盐酸20.27g,调节ph=7,用20g二氯甲烷萃取两次, 合并有机相,无水硫酸钠干燥,减压浓缩得白色固体,经测定:
[0060]
核磁(bruker公司,400m hz,下同)数据如下:
[0061]1h nmr(cdcl3,δ):1.42(6h,s),2.54(2h,s)
[0062]
质谱(质谱测定仪器购自日本岛津公司,型号为lcms-8040,下同) 数据如下:esi:
131
[0063]
hplc检测纯度为98.8%,收率为94.2%。
[0064]
实施例2
[0065]
向反应四口瓶中加入正丁醇26.6g、硫脲8.4g搅拌升温至60~64℃,加 入式(i)所示化合物(x为氯)13.3g继续反应2hr,加入氢氧化钾12.2g, 60℃反应2.5hr,hplc检测式(i)所示化合物完全转化,减压浓缩蒸除正 丁醇,冷却至室温,加入40g水和36%浓盐酸20.27g,调节ph=7,用20g 二氯甲烷萃取两次,合并有机相,无水硫酸钠干燥,减压浓缩得白色固体, 经测定:
[0066]
核磁数据和质谱数据同实施例1,hplc检测纯度为99.1%,收率为 93.8%。
[0067]
实施例3
[0068]
向反应四口瓶中加入乙腈106.4g、硫脲9.12g搅拌升温至78~80℃,加 入式(i)所示化合物(x为氯)13.3g继续反应2hr,加入氢氧化锂5.98g, 80℃反应2hr,hplc检测式(i)所示化合物完全转化,减压浓缩蒸除乙腈, 冷却至室温,加入40g水和36%浓盐酸24.3g,调节ph=7,用20g二氯甲烷 萃取两次,合并有机相,无水硫酸钠干燥,减压浓缩得白色固体,经测定:
[0069]
核磁数据和质谱数据同实施例1,hplc检测纯度为98.9%,收率为93.1%。
[0070]
实施例4
[0071]
按照实施例1的方法,不同的是,将式(i)所示的化合物、硫脲和碱 的摩尔比设置为1:1:3。
[0072]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.6%,收率为91.1%。
[0073]
实施例5
[0074]
按照实施例1的方法,不同的是,将式(i)所示的化合物、硫脲和碱 的摩尔比设置为1:1:1.5。
[0075]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为82.2%,收率为67.7%。
[0076]
实施例6
[0077]
按照实施例1的方法,不同的是,将反应温度控制为50℃。
[0078]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.5%,收率为88%。
[0079]
实施例7
[0080]
按照实施例1的方法,不同的是,将反应温度控制为23~25℃。
[0081]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为97.5%,收率为78.1%。
[0082]
实施例8
[0083]
按照实施例1的方法,不同的是,将式(i)所示的化合物与所述溶剂 的质量比设置为1:1.5。
[0084]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.3%,收率为87.8%。
[0085]
实施例9
[0086]
按照实施例1的方法,不同的是,将式(i)所示的化合物与所述溶剂 的质量比设置为1:10。
[0087]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.7%,收率为94.1%。
[0088]
实施例10
[0089]
按照实施例1的方法,不同的是,等摩尔量的式(i)所示的化合物中, x为氯替换为:x为溴,有机溶剂替换为异丙醇,且式(i)所示的化合物与 所述溶剂的质量比不变。
[0090]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.6%,收率为93.0%。
[0091]
实施例11
[0092]
按照实施例1的方法,不同的是,等摩尔量的式(i)所示的化合物中, x为氯替换为:x为otf,有机溶剂替换为乙醇,且式(i)所示的化合物与 所述溶剂的质量比不变。
[0093]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.2%,收率为93.1%。
[0094]
实施例12
[0095]
按照实施例1的方法,不同的是,等摩尔量的式(i)所示的化合物中, x为氯替换为:x为oms,有机溶剂替换为叔丁醇,且式(i)所示的化合 物与所述溶剂的质量比不变。
[0096]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.3%,收率为91.5%。
[0097]
实施例13
[0098]
按照实施例1的方法,不同的是,等摩尔量的式(i)所示的化合物中, x为氯替换为:x为ots。
[0099]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.2%,收率为90.7%。
[0100]
实施例14
[0101]
按照实施例1的方法,不同的是,等摩尔量的式(i)所示的化合物中, x为氯替换为:x为ons。
[0102]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.5%,收率为91.5%。
[0103]
实施例15
[0104]
按照实施例1的方法,不同的是,碱替换为等摩尔量的碳酸氢钠。
[0105]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.1%,收率为78.6%。
[0106]
实施例16
[0107]
按照实施例1的方法,不同的是,碱替换为实施例1的0.5倍摩尔量的 碳酸钠。
[0108]
对得到的白色固体进行检测,核磁数据和质谱数据同实施例1,hplc 检测纯度为98.2%,收率为72.7%。
[0109]
由本技术各实施例的结果可知,本技术的方法,简洁、高效,无需引入 催化剂,反
应条件温和、反应速度快、反应过程粗放,对设备要求低,产物 纯度高、收率高、产物易分离,具有很好的工业化应用前景。
[0110]
且本领域技术人员应知,在收率高达75%及以上时,在此基础上将收率 提高至90%,是很有难度的,进一步地,将收率再提高至93%-95%是更明 显的提高和进步。
[0111]
将本技术实施例1与实施例4-实施例5的结果比较可知,式(i)所示 的化合物、硫脲和碱的摩尔比为1:(1-1.2):(2-2.5)时,能提高反应产 物的收率和纯度。
[0112]
将本技术实施例1与实施例6-实施例7的结果比较可知,反应温度控制 为60-80℃时,能提高反应产物的收率和纯度。
[0113]
将本技术实施例1与实施例8-实施例9的结果比较可知,式(i)所示 的化合物与所述溶剂的质量比为1:(2-8)时,能够进一步提高反应产物的 收率和纯度以及溶剂的利用率。
[0114]
将本技术实施例1与实施例15-实施例16的结果比较可知,碱为氢氧化 钠、氢氧化钾和氢氧化锂中的至少一种时,能够进一步提高反应产物的收率 和纯度。
[0115]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实 施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方 案进行多种简单变型,这些简单变型均属于本发明的保护范围。
[0116]
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特 征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必 要的重复,本发明对各种可能的组合方式不再另行说明。
[0117]
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其 不违背本发明的思想,其同样应当视为本发明所发明的内容。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。