1.本发明属于医药化学技术领域,具体涉及一种三取代苯基-1,2,4-三氮唑衍生物及其制备和治疗神经元损伤的应用。
背景技术:
2.神经退行性疾病是一类神经细胞发生退行性病变的神经元损伤疾病,包括脑卒中、阿尔兹海默症、帕金森症和肌萎缩性侧索硬化症等。大量研究表明,当神经系统受到损伤和刺激时,神经细胞的氧化应激通路会出现异常,从而导致细胞内的活性氧自由基(ros)或活性氮自由基(nos)水平过高,进而对细胞内的dna,rna,蛋白质和脂质等生物大分子造成损伤,最终导致神经细胞死亡。其中,氧化应激产生的自由基是导致神经退行性疾病的重要原因和病理特征。因此,设计具有对抗氧化应激作用的化合物是治疗神经退行性疾病的一种有效策略。
3.依达拉奉是一种被广泛研究的自由基清除剂,在临床上主要用于急性缺血性脑卒中的治疗,研究表明依达拉奉对肌萎缩侧索硬化症和阿尔兹海默症等神经退行性疾病也具有一定的治疗作用。此外,有研究表明3,5-二芳基取代的噁二唑类化合物具有良好的抗氧化和抗炎性能,能够在体内和体外发挥神经保护作用。
4.目前,仍有多种神经退行性疾病尚无特效药,当前该领域存在的主要问题是许多药物在开发过程中动物实验效果虽然较好,但临床效果较差,如3,5-二芳基取代的噁二唑,该药物由于极性较大,较难穿越血脑屏障,进而影响了其效果的发挥。因此,有必要开发临床效果好的新型神经元损伤防治药物。
技术实现要素:
5.为了克服上述现有技术的不足,本发明的首要目的是提供一种三取代苯基-1,2,4-三氮唑衍生物。
6.本发明的第二个目的是提供上述三取代苯基-1,2,4-三氮唑衍生物的制备方法。
7.本发明的第三个目的是提供上述三取代苯基-1,2,4-三氮唑衍生物的应用。该化合物具有预防和治疗神经元损伤疾病的潜在作用。
8.本发明的上述第一个目的是通过以下技术方案来实现的:
9.一种三取代苯基-1,2,4-三氮唑衍生物,所述衍生物的结构如式i所示:
10.11.式i中:r1独立地选自-h,-oh,-ch2oh,r(c
1-c4),-or(c
1-c4),-x(f,cl,br),-cx3(f,cl),-no2,-nh2,-nr2(c
1-c4),-cooh,-cor(c
1-c4),-coor(c
1-c4);r2独立地选自-h,-oh,-ch2oh,r(c
1-c4),-or(c
1-c4),-x(f,cl,br),-cx3(f,cl),-no2,-nh2,-nr2(c
1-c4),-cooh,-cor(c
1-c4),-coor(c
1-c4);r3独立地选自-oh,-ch2oh,r(c
1-c4),-or(c
1-c4),-x(f,cl,br),-cx3(f,cl),-no2,-nh2;m分别为0,1,2,3。
12.优选地,r1为cf3,r2为h,r3独立地选自4-ch3,4-ch2ch3,4-f,4-cl,4-br,4-cf3,2-f,2-cl,2-cf3,3,5-f,3,5-cf3,3,5-ch3。
13.优选地,所述衍生物还包括具有式i所示结构的前体或药学上可接受的盐。
14.进一步地,所述前体包括酯,醚等前体药物形式,药学上可接受的盐包括与碱金属所形成的盐(如钠盐,钾盐等)。
15.本发明的上述第二个目的是通过以下技术方案来实现的:
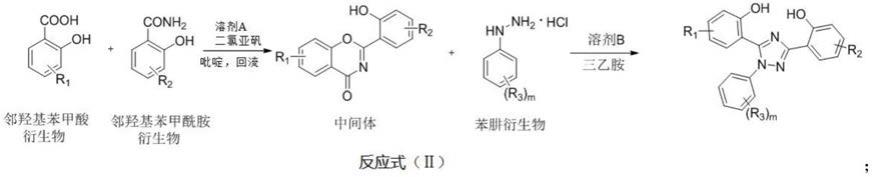
16.上述三取代苯基-1,2,4-三氮唑衍生物的制备方法如反应式(ⅱ)所示:
[0017][0018]
根据反应式(ⅱ),所述制备方法包括以下步骤:
[0019]
s1、将邻羟基苯甲酸衍生物和邻羟基苯甲酰胺衍生物加入溶剂a中,再加入吡啶(催化量),然后边搅拌边加入二氯亚砜,经加热回流后除去溶剂a,再通过溶剂b重结晶得到噁嗪中间体;
[0020]
s2、步骤s1所得的噁嗪中间体和苯肼衍生物加入溶剂b和三乙胺中,经加热回流后除去溶剂b,再通过重结晶得到三取代苯基-1,2,4-三氮唑衍生物。
[0021]
本发明通过将依达拉奉和3,5-二芳基取代的噁二唑为先导化合物,设计出一类新型的三苯基取代的1,2,4-三氮唑类化合物。该类化合物具有三苯基取代的结构,显著提高了化合物的亲酯作用,有利于化合物最终穿越血脑屏障,发挥神经保护作用;两个对称酚羟基及三氮唑杂环有利于清除自由基,发挥抗氧化应激的神经保护作用;苯环上的取代基可以通过电子效应,更好的加强这两种作用。
[0022]
优选地,所述邻羟基苯甲酸衍生物为2-羟基-4-(三氟甲基)苯甲酸,所述邻羟基苯甲酰胺衍生物为2-羟基苯甲酰胺,所述苯肼衍生物包括对甲基苯肼盐酸盐、对乙基苯肼盐酸盐、对氟苯肼盐酸盐、对氯苯肼盐酸盐、对溴苯肼盐酸盐、对三氟甲基苯肼盐酸盐、2-氟苯肼盐酸盐、2-氯苯肼盐酸盐、2-三氟甲基苯肼盐酸盐、2-三氟甲基苯肼盐酸盐、3,5-二三氟甲基苯肼盐酸盐、3,5-二甲基苯肼盐酸盐。
[0023]
优选地,所述溶剂a包括甲苯、二甲苯、二苯醚、n,n-二甲基甲酰胺(dmf),n,n-二甲基乙酰胺,n-甲基吡咯烷酮、二甲亚砜(dmso)中的至少一种。
[0024]
优选地,所述溶剂b包括甲醇、乙醇、丙醇、异丙醇、叔丁醇、四氢呋喃、二氧六环、丙酮、丁酮、乙腈、乙酸乙酯、三氯甲烷、1,1-二氯乙烷、苯、甲苯、吡啶中的至少一种。
[0025]
优选地,所述邻羟基苯甲酸衍生物与邻羟基苯甲酰胺衍生物的物质的量之比为1:(0.8~3),噁嗪中间体与苯肼衍生物的物质的量之比为1:(0.8~4)。
[0026]
优选地,步骤s1的加热回流温度为130-160℃,时间为6-12小时;步骤s2的加热回流温度为60-100℃,时间为6-12小时。
[0027]
优选地,所述邻羟基苯甲酸衍生物与溶剂a的物料比为1:10-50(g/ml)。
[0028]
优选地,所述邻羟基苯甲酸衍生物与二氯亚砜的物料比为1:1-5(g/ml)。
[0029]
优选地,所述噁嗪中间体与溶剂b的物料比为1:10-50(g/ml)。
[0030]
优选地,所述噁嗪中间体与三乙胺的物料比为1:0.1-2(g/ml)。
[0031]
本发明的上述第三个目的是通过以下技术方案来实现的:
[0032]
上述三取代苯基-1,2,4-三氮唑衍生物在制备治疗神经元损伤的药物中的应用。
[0033]
本发明通过利用硝普纳(snp)诱导的pc12细胞损伤模型进行活性筛选,发现本发明的化合物具有良好的抗氧化性能和细胞保护作用,并具有良好的剂量依赖关系;大鼠的大脑动脉闭塞模型实验(mcao)显示,该化合物具有良好的神经保护作用,且该化合物具有预防和治疗神经元损伤疾病的潜在作用。
[0034]
优选地,所述防治神经元损伤的药物的适应症为神经退行性疾病,所述神经退行性疾病包括但不限于帕金森病、阿尔兹海默症、肌萎缩侧索硬化症、脑卒中、脑损伤和脊髓损伤。
[0035]
本发明还提供了一种治疗神经元损伤的药物,所述药物以上述的三取代苯基-1,2,4-三氮唑衍生物为主要活性成分。
[0036]
优选地,在制备所述治疗神经元损伤的药物时,所述三取代苯基-1,2,4-三氮唑衍生物在细胞水平的药物浓度为1-100μm,动物水平的药物浓度为1-60mg/kg。
[0037]
与现有技术相比,本发明的有益效果是:
[0038]
本发明公开了一种三取代苯基-1,2,4-三氮唑衍生物,该化合物以邻羟基苯甲酸衍生物、邻羟基苯甲酰胺衍生物和苯肼衍生物为原料制备而得,该类化合物结构新颖,制备方法简单。同时,该化合物可以显著对抗氧化应激反应,保护神经细胞对抗由硝普钠导致的氧化应激损伤,提高细胞的存活率和神经细胞的形态;同时,该化合物在动物大鼠mcao实验中能够显著减少大脑栓塞体积,对抗缺血再灌注损伤,具有良好的神经保护作用。由于氧化应激是脑卒中、脑损伤、脊髓损伤、阿尔兹海默症、肌萎缩侧索硬化症等疾病的重要病理特征,因此该化合物的优良抗氧化应激作用及血脑屏障穿透作用,可应用于制备预防和治疗脑卒中、脑损伤、脊髓损伤、阿尔兹海默症、肌萎缩侧索硬化症等神经退行性疾病的药物。
附图说明
[0039]
图1为不同硝普纳浓度对pc12细胞存活率的影响(图中为不同浓度的snp处理pc12细胞24小时后的细胞存活率,snp浓度越高,细胞所受到的损伤作用越强,细胞存活率越低);
[0040]
图2为三取代苯基三氮唑衍生物1-12对硝普纳诱导的pc12细胞损伤的保护作用(分别用10μm阳性对照依达拉奉和10μm、5μm、1μm的三氮唑衍生物预处理pc12细胞1小时,然后用400μm snp作用24小时,利用mtt方法测定细胞存活率;与损伤组相比,给药组的细胞存活率得到显著提高,给药浓度越大,存活率提升越明显);
[0041]
图3为化合物12在硝普纳诱导氧化损伤后对pc12细胞形态的维持情况(pc12细胞分别在空白培养基,400μm snp,10μm化合物12和400μm snp作用后,在激光共聚焦显微镜下
位二取代产物)。
[0054]
1、化合物1的合成
[0055]
(1)噁嗪中间体的合成
[0056]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.4g 2-羟基苯甲酰胺加入烧瓶中,加入30ml二甲苯和300μl催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流6h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,用无水乙醇重结晶,得噁嗪中间体。
[0057]
(2)化合物1的合成
[0058]
将1.0g噁嗪中间体和0.5g对甲基苯肼盐酸盐加入烧瓶中,加入15ml无水乙醇和450μl三乙胺,加热回流反应10h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用乙醇重结晶得到目标产物(r1=cf3,r2=h,r3=4-ch3),其结构式如下:
[0059][0060]
目标化合物的结构确证数据:
[0061]
白色固体。1h nmr(500mhz,chloroform-d)δ11.44(s,1h),9.95(s,1h),8.23(d,j=8.1hz,1h),7.39
–
7.37(m,4h),7.36
–
7.31(m,2h),7.27(d,j=8.7hz,1h),7.13(d,j=8.1hz,1h),6.96(d,j=8.3hz,1h),6.66(d,j=7.6hz,1h),2.51(s,3h)。
[0062]
2、化合物2的合成
[0063]
(1)噁嗪中间体的合成
[0064]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.4g 2-羟基苯甲酰胺加入烧瓶中,加入30ml dmf和300μl催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流6h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,用无水乙醇重结晶,得噁嗪中间体。
[0065]
(2)化合物2的合成
[0066]
将1.0g噁嗪中间体和0.6g对乙基苯肼盐酸盐加入烧瓶中,加入15ml无水乙醇和500μl三乙胺,加热回流反应6h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用甲醇重结晶得到目标产物(r1=cf3,r2=h,r3=4-ch2ch3),其结构式如下:
[0067][0068]
目标化合物的结构确证数据:
[0069]
白色固体。1h nmr(500mhz,chloroform-d)δ11.46(s,1h),9.94(s,1h),8.23(d,j=8.1hz,1h),7.44
–
7.38(m,4h),7.36
–
7.31(m,2h),7.27(d,j=8.3hz,1h),7.13(d,j=8.2hz,1h),6.96(d,j=7.9hz,1h),6.65(d,j=7.8hz,1h),2.81(q,j=7.6hz,2h),1.34(t,j=7.6hz,3h)。
[0070]
3、化合物3的合成
[0071]
(1)噁嗪中间体的合成
[0072]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.5g 2-羟基苯甲酰胺加入烧瓶中,加入30ml dmso和300μl催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流6h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,用乙腈重结晶,得噁嗪中间体。
[0073]
(2)化合物3的合成
[0074]
将1.0g噁嗪中间体和0.7g对氟苯肼盐酸盐加入烧瓶中,加入15ml乙腈和450μl三乙胺,加热回流反应7h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用乙腈重结晶得到目标产物(r1=cf3,r2=h,r3=4-f),其结构式如下:
[0075][0076]
目标化合物的结构确证数据:
[0077]
白色固体。1h nmr(500mhz,chloroform-d)δ11.26(s,1h),9.84(s,1h),8.21(d,j=8.1hz,1h),7.54
–
7.48(m,2h),7.38
–
7.26(m,5h),7.14(d,j=9.4hz,1h),6.91(dd,j=8.1,1.6hz,1h),6.68(td,j=8.1,7.7,1.2hz,1h)。
[0078]
4、化合物4的合成
[0079]
(1)噁嗪中间体的合成
[0080]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.6g 2-羟基苯甲酰胺加入烧瓶中,加入30ml二苯醚和300μl催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流7h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,用乙酸乙酯重结晶,得噁嗪中间体。
[0081]
(2)化合物4的合成
[0082]
将1.0g噁嗪中间体和0.6g对氯苯肼盐酸盐加入烧瓶中,加入15ml乙酸乙酯和450μl三乙胺,加热回流反应8h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用乙酸乙酯重结晶得到目标产物(r1=cf3,r2=h,r3=4-cl),其结构式如下:
[0083][0084]
目标化合物的结构确证数据:
[0085]
白色固体。1h nmr(500mhz,chloroform-d)δ11.15(s,1h),9.81(s,1h),8.21(d,j=8.1hz,1h),7.56(d,j=8.6hz,2h),7.46(d,j=8.6hz,2h),7.36(t,j=8.3hz,1h),7.33(s,1h),7.27(d,j=7.5hz,1h),7.14(d,j=8.3hz,1h),6.94(d,j=8.2hz,1h),6.70(t,j=7.6hz,1h)。
[0086]
5、化合物5的合成
[0087]
(1)噁嗪中间体的合成
[0088]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.7g 2-羟基苯甲酰胺加入烧瓶中,加入30ml二甲苯和催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流8h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,用叔丁醇重结晶,得噁嗪中间体。
[0089]
(2)化合物5的合成
[0090]
将1.0g噁嗪中间体和0.7g对溴苯肼盐酸盐加入烧瓶中,加入15ml叔丁醇和450μl三乙胺,加热回流反应10h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用叔丁醇重结晶得到目标产物(r1=cf3,r2=h,r3=4-br),其结构式如下:
[0091][0092]
目标化合物的结构确证数据:
[0093]
白色固体。1h nmr(500mhz,chloroform-d)δ11.13(s,1h),9.80(s,1h),8.21(d,j=8.1hz,1h),7.72(d,j=8.5hz,2h),7.40(d,j=8.8hz,2h),7.36(t,j=7.6hz,1h),7.33(s,1h),7.27(d,j=8.2hz,1h),7.14(d,j=8.3hz,1h),6.95(d,j=7.8hz,1h),6.71(t,j=7.2hz,1h)。
[0094]
6、化合物6的合成
[0095]
(1)噁嗪中间体的合成
[0096]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.8g 2-羟基苯甲酰胺加入烧瓶中,加入30ml dmf和300μl催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流6h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,
用异丙醇重结晶,得噁嗪中间体。
[0097]
(2)化合物6的合成
[0098]
将1.0g噁嗪中间体和1.0g对三氟甲基苯肼盐酸盐加入烧瓶中,加入15ml异丙醇和450μl三乙胺,加热回流反应6h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用丙酮重结晶得到目标产物(r1=cf3,r2=h,r3=4-cf3),其结构式如下:
[0099][0100]
目标化合物的结构确证数据:
[0101]
白色固体。1h nmr(500mhz,chloroform-d)δ10.87(s,1h),9.79(s,1h),8.23(d,j=8.1hz,1h),7.86(d,j=8.3hz,2h),7.68(d,j=8.3hz,2h),7.38(td,j=7.9,1.8hz,1h),7.34(s,1h),7.28(d,j=8.3hz,1h),7.16(d,j=8.5hz,1h),6.92(dd,j=8.0,1.6hz,1h),6.73(t,j=8.0hz,1h)。
[0102]
7、化合物7的合成
[0103]
(1)噁嗪中间体的合成
[0104]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.4g 2-羟基苯甲酰胺加入烧瓶中,加入30mln-甲基吡咯烷酮和300μl催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流7h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,用吡啶重结晶,得噁嗪中间体。
[0105]
(2)化合物7的合成
[0106]
将1.0g噁嗪中间体和0.5g 2-氟苯肼盐酸盐加入烧瓶中,加入15ml吡啶和450μl三乙胺,加热回流反应9h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用乙酸乙酯重结晶得到目标产物(r1=cf3,r2=h,r3=2-f),其结构式如下:
[0107][0108]
目标化合物的结构确证数据:
[0109]
白色固体。1h nmr(500mhz,chloroform-d)δ11.48(s,1h),9.79(s,1h),8.23(d,j=8.1hz,1h),7.68
–
7.62(m,1h),7.59(t,j=7.4hz,1h),7.41(t,j=7.8hz,1h),7.39
–
7.32(m,3h),7.27(d,j=8.4hz,1h),7.13(d,j=8.3hz,1h),6.91(d,j=8.0hz,1h),6.66(t,j=7.6hz,1h)。
[0110]
8、化合物8的合成
[0111]
(1)噁嗪中间体的合成
[0112]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.6g 2-羟基苯甲酰胺加入烧瓶中,加入30ml dmf和300μl催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流8h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,用异丙醇重结晶,得噁嗪中间体。
[0113]
(2)化合物8的合成
[0114]
将1.0g噁嗪中间体和0.6g 2-氯苯肼盐酸盐加入烧瓶中,加入15ml异丙醇和450μl三乙胺,加热回流反应10h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用甲醇重结晶得到目标产物(r1=cf3,r2=h,r3=2-cl),其结构式如下:
[0115][0116]
目标化合物的结构确证数据:
[0117]
白色固体。1h nmr(500mhz,chloroform-d)δ11.62(s,1h),9.82(s,1h),8.25(d,j=8.0hz,1h),7.67(dd,j=8.1,1.5hz,1h),7.65
–
7.57(m,2h),7.57
–
7.53(m,1h),7.37
–
7.32(m,2h),7.29(d,j=8.3hz,1h),7.14(dd,j=8.4,1.2hz,1h),6.78(dd,j=8.1,1.6hz,1h),6.64(td,j=7.7,1.1hz,1h)。
[0118]
9、化合物9的合成
[0119]
(1)噁嗪中间体的合成
[0120]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.7g 2-羟基苯甲酰胺加入烧瓶中,加入30ml二甲苯和300μl催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流8h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,用四氢呋喃重结晶,得噁嗪中间体。
[0121]
(2)化合物9的合成
[0122]
将1.0g噁嗪中间体和0.9g 2-三氟甲基苯肼盐酸盐加入烧瓶中,加入15ml四氢呋喃和450μl三乙胺,加热回流反应6h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用四氢呋喃重结晶得到目标产物(r1=cf3,r2=h,r3=2-cf3),其结构式如下:
[0123][0124]
目标化合物的结构确证数据:
[0125]
白色固体。1h nmr(500mhz,chloroform-d)δ11.73(s,1h),9.74(s,1h),8.24(d,j=8.1hz,1h),8.01(d,j=7.5hz,1h),7.88
–
7.79(m,2h),7.52(d,j=7.5hz,1h),7.39
–
7.24(m,3h),7.13(d,j=8.3hz,1h),6.68
–
6.57(m,2h)。
[0126]
10、化合物10的合成
[0127]
(1)噁嗪中间体的合成
[0128]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.4g 2-羟基苯甲酰胺加入烧瓶中,加入30ml dmso和300μl催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流6h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,用异丙醇重结晶,得噁嗪中间体。
[0129]
(2)化合物10的合成
[0130]
将1.0g噁嗪中间体和0.7g 2-三氟甲基苯肼盐酸盐加入烧瓶中,加入15ml异丙醇和450μl三乙胺,加热回流反应10h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用丙醇重结晶得到目标产物(r1=cf3,r2=h,r3=3,5-f),其结构式如下:
[0131][0132]
目标化合物的结构确证数据:
[0133]
白色固体。1h nmr(500mhz,chloroform-d)δ10.81(s,1h),9.73(s,1h),8.21(d,j=8.1hz,1h),7.40(td,j=7.7,1.5hz,1h),7.34(s,1h),7.27(d,j=8.1hz,1h),7.15(dd,j=8.4,1.1hz,1h),7.13
–
7.05(m,3h),7.00(dd,j=8.0,1.6hz,1h),6.76(td,j=7.6,1.1hz,1h)。
[0134]
11、化合物11的合成
[0135]
(1)噁嗪中间体的合成
[0136]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.4g 2-羟基苯甲酰胺加入烧瓶中,加入30ml二甲苯和300μl催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流10h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取
滤饼,用二氧六环重结晶,得噁嗪中间体。
[0137]
(2)化合物11的合成
[0138]
将1.0g噁嗪中间体和0.9g 3,5-二三氟甲基苯肼盐酸盐加入烧瓶中,加入15ml二氧六环和450μl三乙胺,加热回流反应8h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用二氧六环重结晶得到目标产物(r1=cf3,r2=h,r3=3,5-cf3),其结构式如下:
[0139][0140]
目标化合物的结构确证数据:
[0141]
白色固体。1h nmr(500mhz,chloroform-d)δ10.03(s,2h),8.24(d,j=8.2hz,1h),8.09(s,1h),8.02(s,2h),7.42(t,j=8.5hz,1h),7.35(s,1h),7.29(d,j=8.2hz,1h),7.17(d,j=8.4hz,1h),6.89(d,j=8.1hz,1h),6.77(t,j=7.7hz,1h)。
[0142]
12、化合物12的合成
[0143]
(1)噁嗪中间体的合成
[0144]
将2.1g 2-羟基-4-(三氟甲基)苯甲酸和1.4g 2-羟基苯甲酰胺加入烧瓶中,加入30ml dmso和催化量的吡啶,在室温下边搅拌边加入2ml二氯亚砜,加热回流9h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,有固体析出,过滤,取滤饼,用无水乙醇重结晶,得噁嗪中间体。
[0145]
(2)化合物12的合成
[0146]
将1.0g噁嗪中间体和1.0g 3,5-二甲基苯肼盐酸盐加入烧瓶中,加入15ml无水乙醇和450μl三乙胺,加热回流反应10h,用薄层色谱法对反应进行监测。反应完毕后将反应液自然冷却到室温,用乙醇重结晶纯化得到目标产物(r1=cf3,r2=h,r3=3,5-ch3),其结构式如下:
[0147][0148]
白色固体。1h nmr(500mhz,chloroform-d)δ11.52(s,1h),9.94(s,1h),8.22(d,j=8.1hz,1h),7.36
–
7.31(m,2h),7.27(d,j=8.6hz,1h),7.23(s,1h),7.13(d,j=8.5hz,1h),7.10(s,2h),6.97(d,j=8.1hz,1h),6.65(t,j=7.7hz,1h),2.41(s,6h)。
[0149]
实施例2化合物1至12的亲脂性计算
[0150]
swissadme是一种用于计算小分子的adme参数、药代动力学性质和成药性质的工具(daina a,michielin o,zoete v.swissadme:a free web tool to evaluate pharmacokinetics,drug-likeness and medicinal chemistry friendliness of small molecules[j].scientific reports.2017,7:42717.)在swissadme上导入化合物1-12的结构式,计算各化合物的油水分配系数log p,分子极性表面积和可旋转键等参数,结果如下表1所示。
[0151]
具有良好血脑屏障通透性的化合物一般具有以下化学性质:分子量小于450,log p在2~5之间;分子极性表面积小于氢键供体数目小于3,可旋转键小于8,通过软件计算,并对化合物的血脑屏障通透打分,化合物分数在2-6之间时通常具有良好的血脑屏障通透性。化合物的亲脂性与油水分配系数log p成正相关,化合物1-12的三苯基为较大的平面芳环,具有良好的亲脂性能,log p在4-6之间,综合其它特性,血脑屏障通透性打分在2-3之间,表明其化学结构特点基本符合血脑屏障通透性的要求,初步判断化合物结构可能具有一定的穿越血脑屏障作用,并用其神经保护作用相关。
[0152]
表1化合物1-12的adme参数计算值
[0153][0154]
实施例3化合物1-12对硝普钠诱导的pc12细胞损伤的保护作用研究
[0155]
(1)硝普纳(snp)诱导pc12细胞损伤模型的建立
[0156]
将大鼠肾上腺嗜铬细胞瘤细胞(pc12细胞)置于含有5%胎牛血清和5%马血清的dmem培养基中,在37℃、5%co2的环境下培养。待细胞贴壁生长一段时间后,用胰蛋白酶进行消化,加培养基稀释后用细胞计数板计算细胞数目。然后,以5000个细胞/孔的密度将pc12细胞接种于96孔板中,加入培养基培养24h,弃去旧培养基,对照组加入新的培养基继续培养,实验组加入含有不同snp浓度(200、300、400、500、600μm)的培养基进行培养。12h后,用mtt法测定细胞存活率,细胞存活率=od
各浓度
/od
control
*100%,实验结果重复三次以上,取平均值作为最终细胞存活率。
[0157]
如图1所示,以对照组的细胞存活率为标准(100%)作图,随着硝普纳浓度增大,
pc12细胞的存活率出现明显下降,因为snp产生大量的活性氧和活性氮,破坏了维持细胞生长所必须的酶或其它蛋白质,导致细胞损伤。
[0158]
(2)化合物1-12的保护作用研究
[0159]
用dmso分别溶解阳性药物依达拉奉和化合物1-12配成10mm的储备液,再用培养基将依达拉奉稀释至100、50、25μm,并用培养基稀释药物(化合物1-12)的测试浓度至10、5、1μm。以5000个细胞/孔的密度将pc12细胞接种于96孔板中,加入培养基,在37℃、5%co2的环境下培养24h,弃去旧培养基,对照组和模型组加入空白培养基,实验组则加入含药培养基预孵24h,然后加入500μm snp作用24h,用mtt法测定细胞存活率。
[0160]
结果如图2所示,在给药浓度为1μm时,化合物就开始对pc12细胞显示出保护作用,提高细胞存活率。其中,化合物1,6,8,9,10和12保护作用较好,能够将细胞存活率提高10%以上。在给药浓度为5μm和10μm时,化合物能对pc12细胞发挥显著的保护作用。其中,化合物1,2,6,8,10,12能够将细胞存活率从60%提升到90%以上。相比之下,依达拉奉阳性对照组的细胞存活率只有70.3%。可见,本发明的化合物具有更优的保护作用。其中,在化合物1-12中,化合物12的保护作用最好,在5μm和10μm时能够将细胞存活提升到100%以上。总的来说,化合物1-12能够在一定程度上保护pc12细胞免受snp造成的损伤,使细胞存活率升高,并且随着化合物浓度升高,细胞保护作用越明显,其药效比依达拉奉更优。而化合物12的药效最好,下面以该化合物为例,进一步评价其抗神经元损伤的作用。
[0161]
实施例4化合物12在硝普钠诱导损伤模型中对细胞的保护作用研究
[0162]
用dmso溶解化合物12配成10mm的储备液,再用培养基稀释至药物测试浓度10μm。将pc12细胞接种于中激光共聚焦皿中,加入培养基,在37℃、5%co2的环境下培养24h。弃去旧培养基,对照组和模型组加入空白培养基,实验组则加入含药培养基预孵1h,再在模型组和实验组中加入snp,使其终浓度为500μm,继续培养24h。弃去培养基,每个孔加入含0.2%hoechst 33258的无血清培养液,在37℃下孵育30min。吸去上清,用空白培养基洗涤细胞2次,再加入一定量空白培养基,在激光共聚焦显微镜下观察不同组细胞的形态学变化。
[0163]
结果如图3所示,对照组的细胞呈长梭形,而且在hoechst 33258染色后细胞核处呈暗蓝色。模型组中,pc12细胞在经过snp诱导的损伤后发生细胞质萎缩,形态由长梭形变成为圆形,染色后细胞核处显示亮蓝色,表明细胞受损伤后发生凋亡,产生核固缩。在实验组中,与模型组相比,细胞形态与对照组相似,呈长梭形,在hoechst 33258染色后细胞核处呈暗蓝色,表明化合物能够在一定程度上保护pc12细胞免受snp诱导产生的凋亡。
[0164]
实施例5化合物12对硝普钠诱导产生的ros的清除作用研究
[0165]
用dmso溶解化合物12配成10mm的储备液,再用培养基稀释至药物测试浓度10μm。将pc12细胞接种于中激光共聚焦皿中,加入培养基,在37℃、5%co2的环境下培养24h。弃去旧培养基,对照组和模型组加入空白培养基,实验组则加入含药培养基预孵1h,再在模型组和实验组中加入snp,使其终浓度为500μm,继续培养24h。弃去培养基,每个孔加入1ml含0.2%dcfh-da的无血清培养液,在37℃下孵育30min。吸去上清,用空白培养基洗涤细胞2次,再加入一定量空白培养基,在激光共聚焦显微镜下观察不同组细胞内的ros变化情况。
[0166]
结果如图4所示,ros可以将dcfh-da氧化成二氯荧光素从而产生绿色的荧光。其中,对照组的荧光信号比较弱,因为细胞中的ros水平较低,仅有部分dcfh被氧化。模型组中,由于加入了硝普纳,导致细胞内产生了大量的ros,所以在激光共聚焦显微镜观察下绿
色荧光显著增强。而实验组显示,加入化合物12能够明显地降低硝普纳所引起的绿色荧光强度,说明化合物12具有良好的ros清除作用。
[0167]
实施例6化合物12对缺血再灌注神经损伤的保护作用研究
[0168]
(1)化合物12注射剂的制备
[0169]
1)蓖麻油注射液
[0170]
配方:化合物12(主药)500mg;聚氧乙烯蓖麻油聚合物(助溶剂)20ml;注射用水80ml;
[0171]
制备工艺:在配制容器中加入80ml的注射用水和20ml的蓖麻油,混合均匀,加入500mg化合物,水浴加热至50℃,搅拌使全部溶解,过滤至澄明,灌封。
[0172]
2)环糊精注射液
[0173]
配方:化合物12(主药)500mg;被他羟丙基环糊精(助溶剂)25ml;注射用水75ml;
[0174]
制备工艺:在配制容器中加入75ml的注射用水和25ml的蓖麻油,混合均匀,加入500mg化合物,水浴加热至50℃,搅拌使全部溶解,过滤至澄明,灌封。
[0175]
(2)分组:对照组(假手术组,sham),采取相同的手术流程但不进行缺血再灌注;模型组(model),进行脑内缺血再灌注损伤;给药组:低剂量(化合物12,3mg/kg),中剂量组(化合物12,10mg/kg),高剂量组(化合物12,30mg/kg);阳性对照组(edaravone,10mg/kg),给予依达拉奉作为阳性对照,溶媒为20%的聚氧乙烯蓖麻油聚合物水溶液。
[0176]
(3)实验步骤:将大鼠麻醉后,在其颈部正中切口,分离左侧颈总动脉,颈外动脉,颈内动脉。在颈内动脉近端扎线,远端放置止血夹后,切一小口,插入尼龙线,将拴线慢慢推入颈内动脉,到达大脑前动脉,阻断颅内血流量,进行缺血损伤。对颈部切口进行消毒,缝合切口,1.5h后稍稍拔出栓线,恢复脑内供血,进行缺血再灌注过程。在缺血前1h和再灌注后4h进行腹腔注射给药,手术后24小时参照zea longa方法(longa e z,weinstein p r,carlson s,et al.reversible middle cerebral artery occlusion without craniectomy in rats.[j].stroke;a journal of cerebral circulation,1989,20:84.)对小鼠进行神经学评分,评分结束后处死动物,取大脑切片进行ttc染色观察。
[0177]
(3)实验结果:ttc染色能够将动物大脑切片的缺血区域染成白色,将正常区域染成红色。zea longa神经学评分方法中,分数越高说明神经学功能损伤越严重。结果如图5所示,与假手术组相比,模型组的脑缺血面积明显增大,达到24.26
±
6.17%,说明经历缺血再灌注后大脑损伤比较严重。此外,模型组的神经评分升高2.06
±
0.32,说明经历缺血再灌注损伤后大鼠的神经功能下降。与模型组相比,给药组的大脑缺血面积明显减小,给药3mg时缺血面积减小到16.37
±
6.51%,给药10mg时缺血面积减小到14.49
±
5.62%,给药30mg时缺血面积减小到12.23
±
8.50%,其效果与阳性药物依达拉奉相当(12.77
±
5.82%)。上述神经学评分实验显示,给予低、中、高剂量的化合物12后,神经学评分相应降低,说明化合物12能够帮助改善脑缺血损伤动物的行为。可见,化合物12对大脑的缺血再灌注损伤具有一定的保护作用,适用于防治神经元损伤疾病。
[0178]
实施例7化合物12对血清中氧化相关生化指标的作用
[0179]
在大脑动脉闭塞模型实验(mcao)实验中,对实验动物进行腹腔静脉取血,分离血清,利用试剂盒检测血清指标超氧化物歧化酶(sod)和丙二醛(mda)的水平。
[0180]
结果如图6所示,与假手术组相比,模型组的sod水平降低(1.14
±
0.17u/ml),mda
水平明显增高(42.78
±
5.62μm),表明动物体内的氧化应激水平上升。给药组中,受试动物血清的sod水平显著升高,其中,中剂量组(1.73
±
0.11u/ml)和高剂量组(1.78
±
0.27u/ml)的药效与依达拉奉(1.80
±
0.09u/ml)相近。可见,给予化合物12防治能够降低受试动物血清的mda水平,其中,低、中、高剂量组均能将血清中的mda水平降低到24μm左右,表明化合物12在动物体内能发挥良好的抗氧化作用。
[0181]
以上对本发明的实施方式作了详细说明,但本发明不限于所描述的实施方式。对于本领域的技术人员而言,在不脱离本发明原理和精神的情况下,对这些实施方式进行多种变化、修改、替换和变型,仍落入本发明的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。