1.本发明属于生物工程技术领域,尤其是涉及一种高产纤维二糖酶菌株及其构建方法与应用。
背景技术:
2.纤维质原料中纤维素、半纤维素、木质素分别占30-40%、15-30%、10-20%。纤维素作为植物光合作用的主要多糖类产物,是地球上最为丰富的可再生性天然资源。据估计,地球上纤维素中所蕴藏的能量大约相当于6400亿吨的石油所含的能量,而且纤维素的可再生性是石油等矿质能源所不可比拟的。纤维素乙醇作为一个极具发展潜力的产业,越来越受到国内外的关注和重视。纤维素乙醇的生产过程主要有原料预处理、水解、发酵及蒸馏。其中,充分利用纤维素酶对纤维素进行水解,有可能从根本上解决人类所面临的能源与粮食问题,其深远影响是不可低估的。
3.纤维素酶是一种多组分的复合酶,包括内切型葡聚糖酶(ec3.2.1.4,也称cx酶、cmc酶)、外切型葡聚糖酶(ec3.2.1.91,也称c1酶、微晶纤维素酶)和纤维二糖酶(ec3.2.21,也称β-葡萄糖苷酶)等3种主要组分[henrissat et al.,1989;beguin,1990]。有关纤维素的酶水解机理至今仍未完全研究清楚,但普遍认为:天然纤维素水解成葡萄糖的过程须依靠这三种主要组分的协同作用才能完成。纤维素大分子首先在c1酶和cx酶作用下逐步降解成纤维二糖,而纤维二糖酶则进一步将纤维二糖水解成2个葡萄糖(李宪臻,1996)。
[0004]
目前应用最广、研究最为深入的纤维素酶生产菌种大多是里氏木霉trichoderma reesei的优良突变株。这些菌株能产生高活力的内切型及外切型β-葡聚糖酶,但其纤维二糖酶的含量和活力很低(李宪臻,1996;himmel,1996)。因此,在利用t.reesei的纤维素酶水解纤维素的过程中,常常由于纤维二糖酶的不足造成纤维二糖的积累,而纤维二糖又会对纤维素酶的催化作用形成强烈的反馈抑制(holtzapple et al,1990)。
[0005]
因此,增强纤维二糖酶活力是提高纤维素酶水解得率和葡萄糖产量的关键措施之一。有研究表明在纤维素酶的反应体系中补加纤维二糖酶或利用固定化纤维二糖酶的协同作用可以明显提高纤维素的糖化效率。然而如何得到高产纤维二糖酶的菌株却是一个难点。
[0006]
中国专利cn102994399b公开了一株高产β-葡萄糖苷酶的黑曲霉优势菌株及其应用,具体为黑曲霉aspergillus nigerc112,于2012年4月23日保藏于《中国典型保藏物中心》,其保藏号为cctccno:m 2012129。该发明的黑曲霉aspergillus nigerc112为紫外诱变后,经初筛、复筛后,获得的遗传稳定的突变菌株。通过适当延长黑曲霉aspergillus nigerc112种子培养时间,以麸皮、酵母粉等为原料,经液态发酵,可使β-葡萄糖苷酶酶活达到10iu/ml以上。
[0007]
中国专利cn113403207b公开了一株高产β-葡萄糖苷酶黑曲霉菌株及应用。本发明提供的黑曲霉菌株的保藏号为:cgmcc no.22465,是通过发酵法生产的β-葡萄糖苷酶活力高达88iu/ml,通过与里氏木霉纤维素酶制剂简单复配后形成复合纤维素酶制剂,复合纤维
素酶制剂酶系更加平衡,生物质高固酶解条件下,葡萄糖释放量均提高35%以上,生物质材料中纤维素转化率》90%以上,并在秸秆单细胞蛋白中完成应用,实现秸秆单细胞蛋白产品中蛋白含量》25%。
[0008]
以上两个专利分别介绍了不同的高产β-葡萄糖苷酶黑曲霉菌株。
技术实现要素:
[0009]
为了提高纤维素酶水解得率,本发明提供一种高产纤维二糖酶菌株及其构建方法与应用
[0010]
本发明的目的可以通过以下技术方案来实现:
[0011]
本发明首先提供一种启动子ppgda基因,其核苷酸序列如seq id no.1所示。
[0012]
本发明还提供一种纤维二糖酶编码基因(cb编码基因),其核苷酸序列如seq id no.2所示。
[0013]
本发明还提供一种重组质粒pgpda-hph,所述重组质粒pgpda-hph是将ppgda基因连接到pgpd-hph质粒上获得的。
[0014]
本发明还提供一种重组质粒pgpda-hph-cb,所述重组质粒pgpda-hph-cb是将所述纤维二糖酶编码基因连接到重组质粒pgpda-hph上获得的。
[0015]
本发明还提供一种ppk2-cb重组质粒,所述ppk2-cb重组质粒是将ppk2质粒用hindⅲ酶切,然后去磷酸化,得到的线性化ppk2质粒与来自pgpda-hph-cb质粒的包含启动子ppgda基因、hph潮霉素基因及cb基因的组合元件进行连接获得的。其中hph潮霉素基因核苷酸序列如seq id no.3所示。
[0016]
在本发明的一个实施方式中,所述启动子ppgda基因、hph潮霉素基因及cb基因的组合元件通过以下方法获得:
[0017]
以重组质粒pgpda-hph-cb为模版,设计引物,扩增得到所述启动子ppgda基因、hph潮霉素基因及cb基因的组合元件,其中,
[0018]
上游引物:
[0019]
ppk2-gpda-f:5
’‑
catctccactcgacctgcaggcatgcaagctttgtgacgaactcgtgagctc-3’[0020]
下游引物:
[0021]
gpda-cb-r:5
’‑
gacgttgtaaaacgacggccagtgccaagctttcagactctattgggatacc-3’。
[0022]
本发明还提供一种农杆菌菌株,所述农杆菌菌株是将ppk2-cb重组质粒转化到农杆菌中而获得的新菌株。
[0023]
本发明还提供一种高产纤维二糖酶菌株,为将所述纤维二糖酶编码基因整合到里氏木霉以后所得的一种新的里氏木霉。
[0024]
本发明还提供所述高产纤维二糖酶菌株的构建方法,包括以下步骤:
[0025]
1)、将启动子ppgda基因和纤维二糖酶编码基因cb构成一个表达元件整合到pgpd-hph载体上;
[0026]
2)、将ppgda基因、cb基因和hph潮霉素基因的组合元件整合到ppk2载体上;
[0027]
3)、利用根癌农杆菌介导里氏木霉转化并获得高产纤维二糖酶的里氏木霉工程菌株,即所述高产纤维二糖酶菌株。
[0028]
本发明还提供一种高产纤维二糖酶菌株的应用,利用高产纤维二糖酶菌株水解纤维素。
[0029]
本发明以高产纤维二糖酶的黑曲霉菌株为来源菌株,提取总rna并以此为模板,用rt-pcr法快速扩增了纤维二糖酶cdna,对其全长进行了序列测定并与文献报道的基因序列进行了比较,将其克隆到表达载体ppk2中表达。
[0030]
与现有技术相比,本发明将纤维二糖酶基因整合到里氏木霉中,大幅提高了里氏木霉中纤维二糖酶活力(具体而言,纤维二糖酶酶活提高了273%),改善了里氏木霉的纤维素酶系,从而使可再生资源的酶法糖化得到长足的进展。
附图说明
[0031]
图1.ppgda基因琼脂糖凝胶电泳图;
[0032]
图1中:m为maker条带,1-5为ppgda基因条带;
[0033]
图2.pgpda-hph质粒构建图谱;
[0034]
图3.菌液pcr琼脂糖凝胶电泳图;
[0035]
图3中:m为maker条带,1-4为ppgda基因条带;
[0036]
图4.黑曲霉总rna琼脂糖凝胶电泳图;
[0037]
图5.cb基因琼脂糖凝胶电泳图;
[0038]
图5中,m为maker条带,1、2为cb基因条带;
[0039]
图6.菌液pcr琼脂糖凝胶电泳图;
[0040]
图6中,m为maker条带,1为cb基因条带;
[0041]
图7.重组质粒pgpda-hph-cb图谱;
[0042]
图8.启动子ppgda基因、hph潮霉素基因及cb基因组合元件;
[0043]
图8中,1-5为基因组和表达元件条带,m为maker条带;
[0044]
图9.ppk2-cb质粒酶切验证结果;
[0045]
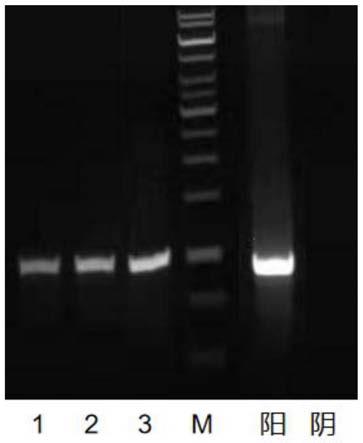
图10.根癌农杆菌转化子菌液pcr验证结果;
[0046]
图10中,m为maker条带,1-5为根癌农杆菌转化子,阳为ppk2-cb质粒阳性对照,阴为空载的农杆菌阴性对照;
[0047]
图11.里氏木霉与根癌农杆菌共培养;
[0048]
图12.倒扣揭膜后转化子生成情况;
[0049]
图13.里氏木霉转化子基因组中cb基因验证;
[0050]
图13中,m为maker条带,1、2、3为里氏木霉转化子,ppk2-cb质粒为阳性对照,里氏木霉出发菌株为阴性对照。
具体实施方式
[0051]
下面结合附图和具体实施例对本发明进行详细说明。
[0052]
实施例1
[0053]
本实施例提供一种高产纤维二糖酶菌株的构建方法,包括以下步骤:
[0054]
1、将启动子ppgda基因和纤维二糖酶编码基因cb构成一个表达元件整合到pgpd-hph载体上;
[0055]
2、将ppgda基因、cb基因和hph潮霉素基因组合元件整合到ppk2载体上;
[0056]
3、利用根癌农杆菌介导里氏木霉转化并获得高产纤维二糖酶的里氏木霉工程菌株。
[0057]
本实施例的具体实验方法如下:
[0058]
1、ppgda启动子的获取
[0059]
以ppk2质粒为模版,设计引物进行pcr,通过1%琼脂糖凝胶电泳验证。
[0060]
上游引物ppgda-f:
[0061]
5'-acctccccacatcacagaaatcaaaactagttgtgacgaactcgtgagctc-3'(下划线为spe i酶切位点)
[0062]
下游引物ppgda-r:
[0063]
5'-agtaacgttaagtggatccgaattcgatatcggtgatgtctgctcaagcgg-3'(下划线为ecor v酶切位点)
[0064]
pcr反应体系:dna mix 10μl,上、下游引物(10ng/μl)各0.4μl,dna模版(10ng/μl)0.2μl,dmso 0.8μl,灭菌水8.2μl,总体积为20μl。
[0065]
pcr反应条件:95℃预变性5min,95℃变形30s,50℃退火1min,72℃延伸10min,循环20次,72℃再延伸20min,4℃保存。反应结束后将pcr产物进行1%琼脂糖电泳。
[0066]
ppgda基因琼脂糖凝胶电泳图如图1所示,由图1可知,在1000bp附近有一条特异性条带,为ppgda基因(903bp)片段位置。启动子ppgda基因核苷酸序列如seq id no.1所示。
[0067]
2、ppgda基因pcr产物回收及纯化
[0068]
采用neb公司dna gel extraction kit柱式dna胶回收试剂盒回收pcr产物,具体操作步骤参照试剂盒中的方法进行操作,回收的dna样品于-20℃贮存备用。
[0069]
3、pgpd-hph质粒酶切和启动子重组质粒pgpda-hph的构建
[0070]
对pgpd-hph质粒进行spe i和ecor v双酶切,然后采用gibson组装的方法将ppgda基因连接到pgpd-hph载体上,得到新的重组质粒pgpda-hph。
[0071]
其中pgpd-hph质粒为已知质粒。
[0072]
图2为pgpd-hph质粒与启动子ppgda进行连接组装得到重组质粒pgpda-hph。
[0073]
双酶切体系:spe i和ecor v酶各1.5μl,cut smart buffer 5μl,dna 2μg(依浓度计算)水补齐至50μl,37℃,3h。
[0074]
gibson连接体系:酶6μl,载体和片段共4μl,比例为1:3~5,金属浴50℃,1h。
[0075]
4、转化到大肠杆菌感受态dh5α中
[0076]
将上一步中的10μl连接产物-重组质粒pgpda-hph与100μl的大肠杆菌dh5α感受态细胞混合,冰浴30min,42℃热激90s,立即重置于冰上3-5min,再加入800μl lb液体培养基,37℃培养50-60min;涂布于lb固体平板(含10μg/m l氨苄青霉素),37℃培养过夜。
[0077]
5、菌液pcr初步鉴定
[0078]
用移液抢挑取转化子单克隆至lb液体培养基中培养6-8h后,进行菌液pcr。
[0079]
上游引物f:5'-tgtgacgaactcgtgagctc-3'
[0080]
下游引物r:5'-ggtgatgtctgctcaagcgg-3'
[0081]
菌液pcr体系:酶5μl,上、下游引物各0.5μl,菌液0.5μl,灭菌水3.5μl总体积为10μl。
[0082]
菌液pcr过程:预变性94℃10min,变性94℃30s,退火55℃30s,延伸72℃2min,再延伸72℃10min,30个循环,4℃保存。反应结束后将pcr产物进行1%琼脂糖电泳检测验证,验证正确的菌落。
[0083]
菌液pcr琼脂糖凝胶电泳图如图3所示,由图3可知,通过菌液pcr验证,存在ppgda基因条带,说明重组质粒成功转化到dh5α中。
[0084]
6、重组质粒pgpda-hph测序。
[0085]
将菌落pcr鉴定为阳性克隆的转化子进行5mllb液体试管培养12-16h,并送往擎科生物(北京)股份有限公司进行dna测序;将测序后获得的序列用dnaman软件进行比对以验证序列的正确性和该方法的可靠性。测序结果表明,所获得的dna片段含有启动子ppgda基因,说明重组质粒构建成功。
[0086]
7、重组质粒pgpda-hph的小量提取
[0087]
采用neb公司plamidminiprepkit试剂盒小量提取质粒,具体操作步骤参照试剂盒中的方法进行操作。提取的pgpda-hph重组质粒于-20℃保存备用。
[0088]
8、黑曲霉总rna提取
[0089]
本实施例以黑曲霉菌株为纤维二糖酶基因来源菌株,所用黑曲霉菌株为购自于中国普通微生物菌种保藏管理中心(cgmcc)菌种编号为cgmcc3.7206的黑曲霉(aspergillusniger)。
[0090]
采用聚合美公司的rna提取试剂盒提取,具体操作步骤参照试剂盒中的方法进行操作。提取的rna于-80℃保存备用。
[0091]
黑曲霉总rna琼脂糖凝胶电泳图如图4所示。rna提取一般显示三条带,分别为28s、18s和5s。图4中有清晰的三条带,说明rna提取的较好。
[0092]
9、构建黑曲霉cdna文库
[0093]
采用聚合美快速反转录试剂盒获取黑曲霉cdna,具体操作步骤如下:
[0094]
(1)根据以下表格在冰上配制反应体系,总体积为10μl。
[0095]
5xsprintgdnaremovermix2μl
[0096]
rna模板0.01~1μg
[0097]
depc-ddh2o补足至10μl
[0098]
将反应液轻轻搅拌均匀,短暂离心使管壁上的溶液收集到管底,42℃温育2分钟,然后置于冰上冷却。
[0099]
(2)接着反转录反应,在冰上加入下列成分:
[0100]
步骤(1)的反应液10μl
[0101]
5xm5sprintrtmix4μl
[0102]
depc-ddh2o6μl
[0103]
(3)轻轻混匀,短暂离心;50℃孵育,5min;85℃加热5秒钟使酶失活;置于冰上进行后续实验或冷冻保存。
[0104]
10、扩增纤维二糖酶编码基因(cb)
[0105]
以步骤9中反转录获得的cdna为模版,设计引物扩增cb基因,通过1%琼脂糖凝胶电泳验证。
[0106]
pcr体系和程序同步骤1。
[0132]
下游引物:
[0133]
gpda-cb-r:5
’‑
gacgttgtaaaacgacggccagtgccaagctttcagactctattgggatacc-3’[0134]
所得产物为启动子ppgda基因、hph潮霉素基因及cb基因的组合元件。
[0135]
图8为启动子pgpda基因及cb基因组合元件,通过pcr及电泳验证,确定得到了所需的基因组合元件。
[0136]
17、ppk2线性化载体与ppk2-cb重组质粒构建
[0137]
ppk2质粒用hindⅲ酶切,然后去磷酸化,方法参照步骤11。得到的线性化ppk2质粒与步骤16中的启动子ppgda基因、hph潮霉素基因及cb基因的组合元件进行gibson组装,得到ppk2-cb重组质粒,方法参照步骤11。
[0138]
18、将重组质粒ppk2-cb转化到大肠杆菌中
[0139]
具体操作同步骤4。
[0140]
19、酶切验证
[0141]
用hindⅲ内切酶对重组质粒ppk2-cb进行酶切,电泳结果如图9所示,可以看出,ppk2-cb质粒中有两个hindⅲ内切酶位点,内切酶将质粒切成两个基因片段,经电泳验证条带大小正确。。
[0142]
20、将重组质粒ppk2-cb转化到根癌农杆菌agl-1中
[0143]
(1)取-80℃保存的根癌农杆菌agl-1感受态于室温或手心片刻待其部分融化,处于冰水混合状态时插入冰中。
[0144]
(2)每100μl感受态加入0.01-1μg ppk2-cb重组质粒dna,用手拨打管底混匀,依次于冰上静置5分钟、液氮5分钟、37℃水浴5分钟、冰浴5分钟。
[0145]
(3)加入700μl无抗生素的lb或yeb液体培养基,于28℃振荡培养2-3小时。6000rpm离心1分钟收菌,留取100μl左右上清轻轻吹打重悬菌块涂布于含相应抗生素的lb或yeb平板上,倒置放于28℃培养箱培养2-3天。
[0146]
得到含有载体质粒的农杆菌菌株agl1。
[0147]
21、菌液pcr验证
[0148]
将步骤20所得的菌液进行pcr验证,验证是否存在cb基因,同时以ppk2-cb质粒作为阳性对照,以agl-1原始菌株为阴性对照。根癌农杆菌转化子菌液pcr验证结果如图10所示。
[0149]
22、里氏木霉孢子液制备
[0150]
将新鲜培养的里氏木霉(trichoderma reesei)(保藏编号为cgmcc no.17798,在中国专利cn113151264a中已经公开)用孢子液将孢子洗下,稀释到106个/ml,备用,以作为出发菌株。
[0151]
23、根癌农杆菌培养
[0152]
(1)将含有载体质粒的农杆菌菌株agl1在含有50μg/ml卡那霉素和50μg/ml利福平的yeb液体培养基中28℃振荡培养过夜,转速为200rpm;
[0153]
(2)取1.5m1过夜培养的农杆菌培养菌液,2400g,离心5min,im液体洗涤两次接种到5ml含有乙酞丁香酮的诱导培养基(200μm乙酰丁香酮as)中,28℃,200rpm振荡培养,直到达到期望的od 600nm下的0.8左右。
[0154]
24、根癌农杆菌介导转化里氏木霉
[0155]
(1)将100μl经诱导后的农杆菌培养菌液与100μl里氏木霉孢子萌发液(培养时间6h,计数板显微镜下计数约1.0
×
106个/ml)混合,转移到覆盖有大小适中经灭菌后的玻璃纸,含有200μm乙酰丁香酮的im琼脂培养基上,28℃培养2-3天;
[0156]
(2)为了进一步筛选阳性转化子,含有转化子的滤膜被转移到含有100μg/ml潮霉素和200μm头孢噻肟的pda培养基上,28℃再培养2-3天。
[0157]
25、里氏木霉转化子筛选
[0158]
通过里氏木霉孢子与根癌农杆菌在玻璃纸上共培养,然后将玻璃纸倒扣到pda抗性平板上,获得里氏木霉转化子,转化子再经过传代培养获得单株菌落,即为将纤维二糖酶编码基因整合到里氏木霉以后所得的一种新的里氏木霉。
[0159]
①
共培养阶段(24-26℃,48-72h)
[0160]
②
倒扣揭膜培养阶段(28-30℃,60-72h)
[0161]
③
挑取转化子划线培养阶段
[0162]
示意图如图11-12所示。
[0163]
26、里氏木霉转化子基因组验证
[0164]
提取里氏木霉转化子基因组,验证其中是否存在潮霉素基因hph,同时以ppk2-cb质粒作为阳性对照,以里氏木霉出发菌株(保藏编号为cgmcc no.17798)为阴性对照。图13为里氏木霉转化子基因组中hph基因验证,通过验证,三株里氏木霉转化子中均存在hph基因。
[0165]
27、所得里氏木霉转化子发酵验证
[0166]
种子培养基配方:葡萄糖2.5g,玉米浆粉1.25g,种液营养盐溶液100ml,置于500ml三角瓶,磁力搅拌下充分溶解,用20%氢氧化钠调ph 4.8,122℃灭菌40min。
[0167]
发酵培养基配方:葡糖糖0.5%,纤维素粉2.0%,蛋白胨1%,发酵营养盐溶液2%,微量元素溶液0.2%,ph5.5,装量100ml/500ml三角瓶,122℃灭菌60min,接种量10%,25.0℃,185r/min培养。
[0168]
(1)摇瓶发酵验证
[0169]
通过发酵培养168h,检测滤纸酶活和纤维二糖酶酶活。由结果可知,工程菌株(即上述所得里氏木霉转化子)较对照菌株(保藏编号为cgmcc no.17798的出发菌株里氏木霉)均在产酶能力上有显著提高,其中滤纸酶活提高了6.67%,纤维二糖酶酶活提高了273%。
[0170]
表1.工程菌株与对照菌株摇瓶发酵对比
[0171] 滤纸酶活(u/ml)纤维二糖酶酶活(u/ml)工程菌株72.8832.23对照菌株68.328.64
[0172]
(2)发酵罐发酵验证
[0173]
通过发酵罐发酵192h验证,检测滤纸酶活和纤维二糖酶酶活。由结果可知,工程菌株较对照均在产酶能力上有显著提高,其中滤纸酶活提高了5.97%,纤维二糖酶酶活提高了356%。
[0174]
表2.工程菌株与对照菌株发酵罐发酵对比
[0175] 滤纸酶活(u/ml)纤维二糖酶酶活(u/ml)工程菌株192.52101.76
对照菌株181.6822.32
[0176]
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
