1.本发明总体上涉及制造电子装置的领域,并且更具体地涉及在半导体制造中使用的材料的领域。
背景技术:
2.光致抗蚀剂底层组合物在半导体工业中用作集成电路制造的先进技术节点中的光刻的蚀刻掩模。这些组合物通常用于三层和四层光致抗蚀剂集成方案中,其中将有机或含硅的减反射涂层和可图案化光致抗蚀剂膜层布置在具有高碳含量的底层上。
3.理想的光致抗蚀剂底层材料应该具有某些特定特性:它应该能够通过旋涂工艺浇铸到基底上,应该在加热时热固化,具有低脱气和升华,应该可溶于普通溶剂中以具有良好的旋转筒相容性(spin bowl compatibility),应该具有合适的n&k值以与抗反射涂层一起工作以赋予光致抗蚀剂成像所需的低反射率,并且应该具有高的热稳定性以避免在随后的处理步骤期间被损坏。除了这些要求之外,理想的光致抗蚀剂底层材料必须在基底上旋涂和热固化时提供平坦的膜,其具有形貌和对于光致抗蚀剂底层膜上方和下方的含硅层的足够的干法蚀刻选择性,以便以精确的方式将光图案转移到最终的基底中。
4.可交联酚醛清漆树脂已经用于底层应用。酚醛清漆树脂是一种或多种活化芳香族化合物与另一种选自脂肪族或芳香族羰基化合物、苄基醚、苄基醇、或苄基卤化物的单体的缩聚产物。最广泛研究的酚醛清漆树脂是活化芳香族衍生物与甲醛型或芳香醛共聚单体的缩聚产物。这些树脂已经用于各种光刻组合物中的广泛应用中。一种相似类别的树脂是活化芳香族衍生物与酰氯之间的缩聚产物,其中所得的酮在第二步骤中被还原为苄醇以得到高度可溶的可交联材料。
5.仍然需要新的光致抗蚀剂底层材料,其可以提供诸如以下的特性:改善的可溶性、降低的固化温度、高热稳定性、固化之后的耐溶剂性、改善的间隙填充、以及改善的平坦化。
技术实现要素:
6.提供了一种光致抗蚀剂底层组合物,其包含聚合物,所述聚合物包含具有式(1)的重复单元:
[0007][0008]
其中ar是单环或多环c
5-60
芳香族基团,其中所述芳香族基团包含一个或多个芳香族环杂原子、包含杂原子的取代基、或其组合;r1是氢、取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
2-30
烯基、取代或未取代的c
2-30
炔基、取代或未取代的c
6-30
芳基、取代或未取代的c7-30
芳基烷基、取代或未取代的c
7-30
烷基芳基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基;并且r2是取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
2-30
烯基、取代或未取代的c
2-30
炔基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
7-30
烷基芳基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基,其中r1和r2可以任选地一起形成环。
[0009]
还提供了一种形成图案的方法,所述方法包括:(a)在基底上施加光致抗蚀剂底层组合物的层;(b)将所施加的光致抗蚀剂底层组合物固化以形成光致抗蚀剂底层;以及(c)在所述光致抗蚀剂底层上形成光致抗蚀剂层。
具体实施方式
[0010]
现在将详细参考示例性实施例,其实例在本说明书中展示。就这一点而言,本示例性实施例可以具有不同的形式并且不应该被解释为限制于本文所示的描述。因此,下面仅通过参考附图来描述示例性实施例,以解释本说明书的多个方面。如本文所用,术语“和/或”包括相关列出项中的一个或多个的全部组合。当如“......中的至少一个/种”的表述在元件列表之前时,其修饰整个元件列表并且不修饰列表中的单个元件。
[0011]
如本文所用,术语“一个/一种(a,an)”和“该”不表示数量的限制,并且除非在本文中以其他方式指出或与上下文明显矛盾,否则被解释为包括单数和复数二者。除非以其他方式明确指出,否则“或”意指“和/或”。本文所公开的全部范围包括端点,并且所述端点彼此可独立组合。后缀“(s)”旨在包括其修饰的术语的单数和复数二者,由此包括至少一个该术语。“任选的”或“任选地”意指随后描述的事件或情况可能发生或可能不发生,并且所述描述包括所述事件发生的例子以及其没有发生的例子。术语“第一”、“第二”和类似术语在本文不表示顺序、数量、或重要性,而是用于将一个元件与另一个进行区分。当一个元件被称为是“在”另一个元件“之上”时,它可以与所述另一个元件或可能存在于其间的插入元件直接接触。相反,当一个元件被称为是“直接在”另一个元件“之上”时,不存在插入元件。应当理解,可以在各方面中以任何合适的方式来组合所描述的方面的组件、元件、限制和/或特征。
[0012]
除非另有定义,否则本文使用的所有术语(包括技术和科学术语)均具有与本发明所属领域普通技术人员所通常理解的相同含义。进一步将理解,术语(诸如常用词典中定义的那些)应被解释为具有与其在相关领域和本公开的上下文中的含义一致的含义,并且除非本文明确如此定义,否则将不会被解释为理想化或过于正式的意义。
[0013]
如本文所用,术语“烃基”是指具有至少一个碳原子和至少一个氢原子的有机化合物,其任选地在指示的地方被一个或多个取代基取代;“烷基”是指直链或支链的饱和的烃,其具有指定的碳原子数并且具有为1的化合价;“亚烷基”是指具有为2的化合价的烷基;“羟烷基”是指被至少一个羟基(-oh)取代的烷基;“烷氧基”是指“烷基-o
‑”
;“羧酸基”是指具有式
“‑
c(=o)-oh”的基团;“环烷基”是指具有其中全部环成员是碳的一个或多个饱和环的单价基团;“亚环烷基”是指具有为2的化合价的环烷基;“烯基”是指具有至少一个碳碳双键的直链或支链的单价烃基;“烯氧基”是指“烯基-o
‑“
;“亚烯基”是指具有至少为2的化合价的烯基;“环烯基”是指具有至少一个碳碳双键的环烷基;“炔基”是指具有至少一个碳碳三键
的单价烃基;术语“芳香族基团”代表如文献、特别是iupac 19中定义的芳香性的常规观念,并且是指单环或多环芳香族环系统,其在一个或多个环中包含碳原子,并且任选地可以在一个或多个环中包含一个或多个独立地选自n、o和s的代替一个或多个碳原子的杂原子;“芳基”是指单价的单环或多环芳香族基团,其在一个或多个芳香族环中仅含有碳原子,并且可以包含具有稠合到至少一个环烷基或杂环烷基环上的芳香族环的基团;“亚芳基”是指具有至少为2的化合价的芳基;“烷基芳基”是指已被烷基取代的芳基;“芳基烷基”是指已被芳基取代的烷基;“芳氧基”是指“芳基-o
‑”
;并且“芳硫基”是指“芳基-s
‑”
。
[0014]
前缀“杂”意指所述化合物或基团包含作为代替碳原子的杂原子的至少一个成员(例如,1、2、3、或4或更多个杂原子),其中所述一个或多个杂原子各自独立地选自n、o、s、si、或p;“含杂原子的基团”是指包含至少一个杂原子的取代基;“杂烷基”是指具有1-4个代替碳原子的杂原子的烷基;“杂环烷基”是指具有一个或多个代替碳原子的n、o或s原子的环烷基;“亚杂环烷基”是指具有至少为2的化合价的杂环烷基;“杂芳基”是指具有1至3个具有一个或多个代替碳原子的n、o或s原子作为环成员的单独的环或稠环的芳基;并且“亚杂芳基”是指具有至少为2的化合价的杂芳基。
[0015]
术语“卤素”意指氟(氟代)、氯(氯代)、溴(溴代)、或碘(碘代)的单价取代基。前缀“卤代”意指包含代替氢原子的氟、氯、溴、或碘取代基中一个或多个的基团。可以存在卤基(例如溴和氟)的组合或仅氟基团。
[0016]
符号“*”表示重复单元的结合位点(即,附接点)。
[0017]“取代的”意指所述基团上的至少一个氢原子被另一个基团替代,前提是不超过所指定的原子的正常价。当取代基是氧代(即,=o)时,则碳原子上的两个氢被替代。取代基或变量的组合是可允许的。可以存在于“取代的”位置上的示例性基团包括但不限于,硝基(-no2)、氰基(-cn)、羟基(-oh)、氧代(=o)、氨基(-nh2)、单-或二-(c
1-6
)烷基氨基、烷酰基(诸如c
2-6
烷酰基诸如酰基)、甲酰基(-c(=o)h)、羧酸或其碱金属或铵盐、c
2-6
烷基酯(-c(=o)o-烷基或-oc(=o)-烷基)、c
7-13
芳基酯(-c(=o)o-芳基或-oc(=o)-芳基)、酰胺基(-c(=o)nr2,其中r是氢或c
1-6
烷基)、甲酰胺基(-ch2c(=o)nr2,其中r是氢或c
1-6
烷基)、卤素、巯基(-sh)、c
1-6
烷硫基(-s-烷基)、氰硫基(-scn)、c
1-6
烷基、c
2-6
烯基、c
2-6
炔基、c
1-6
卤代烷基、c
1-9
烷氧基、c
1-6
卤代烷氧基、c
3-12
环烷基、c
5-18
环烯基、具有至少一个芳香族环的c
6-12
芳基(例如苯基、联苯基、萘基等,每个环是取代或未取代的芳香族的)、具有1至3个分开的或稠合的环以及6至18个环碳原子的c
7-19
芳基烷基、具有1至3个分开的或稠合的环以及6至18个环碳原子的芳基烷氧基、c
7-12
烷基芳基、c
4-12
杂环烷基、c
3-12
杂芳基、c
1-6
烷基磺酰基(-s(=o)
2-烷基)、c
6-12
芳基磺酰基(-s(=o)
2-芳基)、或甲苯磺酰基(ch3c6h4so
2-)。当基团是取代的时,指示的碳原子数是基团中的碳原子的总数,除了任何取代基的那些。例如,基团-ch2ch2cn是被氰基取代的c2烷基。
[0018]
如上所指出,仍然需要新的光致抗蚀剂底层材料,其可以提供诸如以下的特性:改善的可溶性、降低的固化温度、高热稳定性、固化之后的耐溶剂性、改善的间隙填充、以及改善的平坦化。
[0019]
在光致抗蚀剂底层材料的聚合物单元中并入羧酸酯基团可以明显地改善材料可溶性并且降低用于固化的交联起始温度,而不显著损害耐蚀刻性和反射率参数。具体地,本文公开的本发明的组合物实现优异的平坦化性能,并且具有可以基于聚合物结构调整的蚀
刻速率。包含含羧酸酯的聚合物的光致抗蚀剂底层组合物可以形成交联和/或可以是可交联的,优选地其中所述聚合物在没有照射的情况下是可交联的。例如,本发明的光致抗蚀剂底层组合物可以包含热酸产生剂并且优选地不包含光酸产生剂。交联可以经由交联剂或通过自交联进行。
[0020]
根据一个实施例,光致抗蚀剂底层组合物包含聚合物,所述聚合物包含具有式(1)的重复单元:
[0021][0022]
其中,在式(1)中,ar是单环或多环c
5-60
芳香族基团,其中所述芳香族基团包含一个或多个芳香族环杂原子、包含杂原子的取代基、或其组合。为了方便起见,所述单环或多环c
5-60
芳香族基团可以在本文中被称为“ar基团”。典型地,一个或多个杂原子独立地可以选自n、o、或s。当c
5-60
芳香族基团是多环时,所述一个或多个环基团可以稠合(诸如萘基等)、直接连接(诸如联芳基、联苯基等)和/或通过杂原子桥接(诸如三苯基氨基或二亚苯基醚)。在一个实施例中,多环芳香族基团可以包含稠合和直接连接的环(诸如联萘基等)的组合。应理解,单环或多环c
5-60
芳香族基团的一个或多个杂原子可以作为代替碳原子的芳香族环成员(例如,亚杂芳基)、作为含杂原子的取代基(例如,羟基取代基)的一个或多个杂原子、或其组合存在。
[0023]
单环或多环c
5-60
芳香族基团可以是取代的或未取代的。示例性的取代基包括但不限于取代或未取代的c
1-30
烷基、取代的或未取代的c
1-30
杂烷基、取代的或未取代的c
3-30
环烷基、取代的或未取代的c
2-30
杂环烷基、取代的或未取代的c
2-30
烯基、取代的或未取代的c
2-30
炔基、取代的或未取代的c
6-30
芳基、取代的或未取代的c
7-30
芳基烷基、取代的或未取代的c
7-30
烷基芳基、取代的或未取代的c
3-30
杂芳基、取代的或未取代的c
4-30
杂芳基烷基、卤素、-or
11
、-sr
12
、或-nr
13r14
,其中r
11
至r
14
各自独立地是氢、取代的或未取代的c
1-30
烷基、取代的或未取代的c
3-30
环烷基、取代的或未取代的c
2-30
杂环烷基、取代的或未取代的c
6-30
芳基、取代的或未取代的c
7-30
芳基烷基、取代的或未取代的c
3-30
杂芳基、或取代的或未取代的c
4-30
杂芳基烷基。
[0024]
在一个实施例中,单环或多环c
5-60
芳香族基团可以是单环或多环c
6-60
亚芳基或单环或多环c
5-60
亚杂芳基。当c
5-60
芳香族基团是单环或多环c
6-60
亚芳基时,至少一个氢原子被取代为如上详述的含杂原子的取代基,诸如-or
11
、-sr
12
、或-nr
13r14
,其中r
11
至r
14
各自独立地是氢、取代或未取代的c
1-30
烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基。优选地,ar是多环c
10-60
亚芳基或多环c
7-60
亚杂芳基。示例性ar基团包括但不限于取代或未取代的咔唑二基、取代的亚苯基、取代的亚联苯基、取代的亚萘基、和取代基的芘基。
[0025]
在一个实施例中,单环或多环c
5-60
芳香族基团可以是被-or
11
、-sr
12
、或-nr
13r14
取
代的单环或多环c
6-60
亚芳基,其中r
11
至r
14
各自独立地是氢、取代或未取代的c
1-30
烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基。例如,单环或多环c
5-60
芳香族基团可以是被羟基取代的单环或多环c
6-60
亚芳基。
[0026]
应理解,当“单环或多环c
6-60
亚芳基”是多环时,碳原子数对于基团是足够的以在化学上是可行的。例如,“单环或多环c
6-60
亚芳基”可以是指“单环c
6-60
亚芳基或多环c
10-60
亚芳基”;或例如“单环c
6-30
亚芳基或多环c
12-60
亚芳基”。
[0027]
应理解,当“单环或多环c
5-60
亚杂芳基”是多环时,碳原子数对于基团是足够的以在化学上是可行的。例如,“单环或多环c
5-60
亚杂芳基”可以是指“单环c
5-60
亚杂芳基或多环c
10-60
亚杂芳基”;或例如“单环c
5-30
亚杂芳基或多环c
12-60
亚杂芳基”。
[0028]
在式(1)中,r1是氢、取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
2-30
烯基、取代或未取代的c
2-30
炔基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
7-30
烷基芳基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基。优选地,r1是氢、c
1-10
烷基、c
1-10
氟烷基、c
6-12
芳基、或c
6-12
氟芳基,其中氢是典型的。
[0029]
在式(1)中,r2是取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
2-30
烯基、取代或未取代的c
2-30
炔基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
7-30
烷基芳基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基。优选地,r2是取代或未取代的c
1-20
烷基、取代或未取代的c
1-20
杂烷基、取代或未取代的c
3-20
环烷基、取代或未取代的c
2-20
杂环烷基、取代或未取代的c
2-20
烯基、取代或未取代的c
2-20
炔基、取代或未取代的c
6-24
芳基、或取代或未取代的c
5-20
杂芳基。
[0030]
任选地,r1和r2可以一起形成环。
[0031]
在一个实施例中,ar基团可以是具有式(2)的基团:
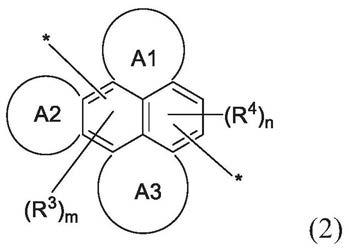
[0032][0033]
其中a1、a2、和a3各自可以是存在或不存在的,并且各自独立地表示1至3个稠合芳香族环。
[0034]
在式(2)中,r3和r4各自独立地是取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
2-30
烯基、取代或未取代的c
2-30
炔基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
7-30
烷基芳基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基、卤素、-or
21
、-sr
22
、或-nr
23r24
,前提是r3或r4中的至少一个是-or
21
、-sr
22
、或-nr
23r24
。
[0035]
在式(2)中,r
21
至r
24
各自独立地是氢、取代或未取代的c
1-30
烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
6-30
芳基、取代或未取代的c7-30
芳基烷基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基。
[0036]
在式(2)中,m式0至4的整数,并且n是0至4的整数,前提是m和n的和是大于0的整数。例如,m和n的和可以是1、2、3、或4或更大,优选地1或2。
[0037]
在另一个实施例中,ar基团可以是具有式(3a)、(3b)、或(3c)的基团:
[0038][0039]
其中a4可以是存在或不存在的,并且表示1至3个稠合芳香族环。优选的是,a4表示1至3个芳香族环,并且更优选地1至2个稠合芳香族环,并且最优选地1个稠合芳香族环。
[0040]
在式(3a)、(3b)、或(3c)中,z1和z2各自独立地是c或n,前提是a4包含至少一个杂芳基环,z1和z2中的至少一个是n,或其组合。
[0041]
在式(3a)、(3b)、或(3c)中,r5各自独立地是取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
2-30
烯基、取代或未取代的c
2-30
炔基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
7-30
烷基芳基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基。在式(3a)、(3b)、和(3c)中,p是0至4的整数,典型地0或1。
[0042]
在另一个实施例中,ar基团可以是具有式(4)的基团:
[0043][0044]
前提是ar基团包含一个或多个芳香族环杂原子、包含杂原子的取代基、或其组合。
[0045]
在式(4)中,l1是单键、-o-、-s-、-s(o)-、-so
2-、-c(o)-、-cr
41r42-、-nr
43
、或-pr
44-,其中r
41
至r
44
各自独立地是氢、取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基。优选地,l1是-o-或-nr
43-,更优选地-nr
43-。
[0046]
在式(4)中,l2不存在、是单键、-o-、-s-、-s(o)-、-so
2-、-c(o)-、取代或未取代的c
1-2
亚烷基、取代或未取代的c
6-30
亚芳基、或取代或未取代的c
5-30
亚杂芳基。优选地,l2是单键。
[0047]
在式(4)中,r8和r9各自独立地是取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
2-30
烯基、取代或未取代的c
2-30
炔基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
7-30
烷基芳基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基、卤素、-or
45
、-sr
46
、或-nr
47r48
。在式(4)中,a是0至4的整数,典型地是0至2,更典型地是
0;并且b是0至4的整数,典型地是0至2,更典型地是0。
[0048]
在式(4)中,r
41
至r
48
各自独立地是氢、取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基。
[0049]
本发明的聚合物可以通过在酸催化剂的存在下并且任选地在合适的溶剂中使一种或多种单环或多环c
5-60
芳香族化合物(芳香族单体)与一种或多种具有式(5)的二羰基化合物(二羰基单体)反应来制备。
[0050][0051]
其中r1是氢、取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
2-30
烯基、取代或未取代的c
2-30
炔基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
7-30
烷基芳基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基。在式(5)中,r2是氢、取代或未取代的c
1-30
烷基、取代或未取代的c
1-30
杂烷基、取代或未取代的c
3-30
环烷基、取代或未取代的c
2-30
杂环烷基、取代或未取代的c
2-30
烯基、取代或未取代的c
2-30
炔基、取代或未取代的c
6-30
芳基、取代或未取代的c
7-30
芳基烷基、取代或未取代的c
7-30
烷基芳基、取代或未取代的c
3-30
杂芳基、或取代或未取代的c
4-30
杂芳基烷基。
[0052]
芳香族单体是单环或多环c
5-60
芳香族化合物,其中所述芳香族化合物包含一个或多个芳香族环杂原子、包含杂原子的取代基、或其组合。示例性c
5-60
芳香族化合物包括但不限于取代的苯、取代的联苯、取代的萘、取代联萘、取代的蒽、取代的苯并[a]蒽、取代的芴、取代的荧蒽、取代的苯并[b]荧蒽、取代的二苯并(a,h)蒽、取代的菲、取代的非那烯、取代的并四苯、取代的取代的苯并菲、取代的芘、取代的并五苯、取代的苯并[a]芘、取代的心环烯、取代的苯并苝、取代的蔻、取代的卵苯、取代的苯并[c]芴、取代或未取代的苯并噻吩、取代或未取代的二苯并噻吩、取代或未取代的咔唑、取代或未取代的吲哚、取代或未取代的喹啉、取代或未取代的异喹啉、取代或未取代的嘌呤、取代或未取代的吩噁嗪、取代或未取代的吩噻嗪、取代或未取代的氧吩噻嗪、取代或未取代的二氧吩噻嗪等。
[0053]
单体和任选的溶剂可以按任何顺序合并。酸催化剂典型地在单体和任何任选的溶剂之后添加到反应混合物中。在添加酸催化剂之后,将反应混合物诸如在回流下典型地加热一段时间,诸如1至48小时。在加热之后,将反应产物诸如通过沉淀从反应混合物中分离,并且典型地干燥,并且任选地纯化,然后再使用。总芳香族单体与总二羰基单体的摩尔比是0.5∶1至2∶1,并且典型地1∶1至1.5∶1。
[0054]
在一个实施例中,聚合物在不使用除具有式(5)的二羰基化合物以外的醛或酮化合物的情况下制备。例如,聚合物不包含来源于具有式ar
′‑
cho的醛的重复单元,其中ar
′
是取代或未取代的c
6-30
芳香族基团。
[0055]
各种溶剂可以用于制备本发明的聚合物,诸如但不限于醇、二醇醚、内酯、酯、醚、酮、水、和芳香族烃。优选地,使用相对极性溶剂,诸如醇、二醇醚、内酯、酯、醚、酮、或水。可
以使用溶剂的混合物。示例性溶剂包括但不限于甲醇、乙醇、丙醇、丙二醇、丙二醇单甲醚(pgme)、丙二醇二甲醚、二乙二醇二甲醚、丙二醇单甲醚乙酸酯(pgmea)、γ-丁内酯(gbl)、γ-戊内酯、6-戊内酯、乳酸乙酯、1,4-二噁烷、环己酮、环戊酮、甲乙酮、水、均三甲苯、二甲苯、苯甲醚、4-甲基苯甲醚等。优选的溶剂是甲醇、乙醇、丙醇、丙二醇、丙二醇单甲醚、丙二醇二甲醚、丙二醇单甲醚乙酸酯、γ-丁内酯、γ-戊内酯、δ-戊内酯、乳酸乙酯、1,4-二噁烷、环己酮、和水。
[0056]
各种酸可以适合地用作本发明的聚合物的制备中的催化剂。示例性酸包括但不限于有机羧酸和二羧酸诸如丙酸和草酸,无机酸和磺酸,并且优选地酸催化剂是无机酸或磺酸。合适的无机酸是hf、hcl、hbr、hno3、h2so4、h3po4、和hclo4。合适的磺酸包括烷烃磺酸和芳基磺酸,诸如甲磺酸、乙磺酸、丙磺酸、苯基磺酸、苯酚磺酸、对甲苯磺酸、和甲酚磺酸。优选的酸催化剂是hcl、hbr、hno3、h2so4、h3po4、甲磺酸、乙磺酸、苯基磺酸、苯酚磺酸、和对甲苯磺酸(ptsa)。
[0057]
在另一个实施例中,所述聚合物可以包含具有式(6)的重复单元:
[0058][0059]
其中r1和r2是如式(1)中所定义的。
[0060]
本发明的聚合物典型地具有500至20000道尔顿(da)、优选地500至15000da、并且更优选地500至10000da的重均分子量(mw),如使用聚苯乙烯标准物通过凝胶渗透色谱法(gpc)确定。
[0061]
光致抗蚀剂底层组合物可以进一步包含溶剂和任选地一种或多种选自固化剂、交联剂、和表面活性剂的添加剂。本领域技术人员将理解,其他添加剂可以适合地在本发明的组合物中使用。
[0062]
溶剂可以是典型地用于电子行业的有机溶剂,诸如pgme、pgmea、3-甲氧基丙酸甲酯(mmp)、乳酸乙酯、乙酸正丁酯、苯甲醚、n-甲基吡咯烷酮、γ-丁内酯(gbl)、乙氧基苯、丙酸苄酯、苯甲酸苄酯、环己酮、环戊酮、碳酸亚丙酯、二甲苯、均三甲苯、枯烯、柠檬烯、及其混合物。典型地,光致抗蚀剂底层组合物的总固体是光致抗蚀剂底层组合物的总重量的0.5wt%至20wt%,典型地0.5wt%至10wt%,光致抗蚀剂底层组合物的剩下部分是溶剂。
[0063]
任选地,本发明的光致抗蚀剂底层组合物可以进一步包含一种或多种固化剂以帮助沉积的聚合物膜的固化。固化剂是引起光致抗蚀剂底层组合物在基底表面上固化的任何组分。优选的固化剂是热酸产生剂(tag)。tag是在暴露于热时释放酸的任何化合物。热酸产生剂在本领域是众所周知的并且通常是如从康涅狄格州诺沃克金氏工业公司(king industries,norwalk,connecticut)可商购的。示例性热酸产生剂包括但不限于胺封端的强酸,诸如胺封端的磺酸,诸如胺封端的十二烷基苯磺酸。本领域技术人员还将理解的是,某些光酸产生剂能够在加热时释放酸并且可以用作热酸产生剂。可用于本发明的组合物的此类固化剂的量可以是例如基于光致抗蚀剂底层组合物的总固体的大于0wt%至10wt%,并且典型地大于0wt%至3wt%。
[0064]
任何合适的交联剂可以用于本发明的组合物,前提是此类交联剂具有至少2个、并且优选至少3个能够在合适的条件下(诸如在酸性条件下)与本发明的聚合物反应的部分。示例性交联剂包括但不限于酚醛清漆树脂、含环氧基的化合物、三聚氰胺化合物、胍胺化合物、含异氰酸酯的化合物、苯并环丁烯、苯并噁嗪等,并且典型地前述中具有2个或更多个、更典型地3个或更多个选自羟甲基、c
1-10
烷氧基甲基、以及c
2-10
酰氧基甲基的取代基的任一项。合适的交联剂的实例是由式(7)和(8)示出的那些。
[0065][0066]
此类交联剂在本领域中是众所周知的并且从多个来源可商购。可用于本发明的组合物的此类交联剂的量可以是例如在基于组合物的总固体的大于0wt%至30wt%,并且典型地大于0wt%至10wt%的范围内。
[0067]
本发明的光致抗蚀剂底层组合物可以任选地包含一种或多种表面流平剂(或表面活性剂)和抗氧化剂。典型的表面活性剂包括展现出两亲性质的那些,意味着它们可以同时既是亲水性的又是疏水性的。两亲性表面活性剂具有一个或多个亲水性头基(其对于水具有强的亲和力)以及一个长疏水尾(其是亲有机性的且排斥水)。合适的表面活性剂可以是离子的(即,阴离子的、阳离子的)或非离子的。表面活性剂的另外的实例包括硅酮表面活性剂、聚(氧化烯)表面活性剂、和含氟化合物表面活性剂。合适的非离子表面活性剂包括但不限于辛基和壬基苯酚乙氧基化物,诸如x-114、x-100、x-45、x-15,以及支链仲醇乙氧基化物,诸如tergitol
tm
tmn-6(陶氏化学公司(the dow chemical company),美国密西根州米德兰)和pf-656(欧诺瓦解决方案公司(omnova solutions),美国俄亥俄州比奇伍德)。还另外的示例性的表面活性剂包括,醇(伯醇和仲醇)乙氧基化物、胺乙氧基化物、葡糖苷、葡糖胺、聚乙二醇、聚(乙二醇-共-丙二醇),或公开于以下中的其他表面活性剂:glen rock,n.j的manufacturers confectioners publishing co.[糖果制造商出版公司]出版的2000年北美版的mccutcheon
′
s emulsifiers and detergents[麦卡琴乳化剂和清洁剂]。作为炔二醇衍生物的非离子表面活性剂也可以是合适的。此类表面活性剂可商购于宾夕法尼亚州阿伦敦的空气化工产品有限公司(air products and chemicals,inc.)并且以商品名和出售。另外合适的表面活性剂包括其他聚合物化合物,诸如三嵌段eo-po-eo共聚物25r2、l121、l123、l31、l81、l101和p123(巴斯夫公司(basf,inc.))。如果使用的话,此类表面活性剂可以基于光致抗蚀剂底层组合物的总固体的例如大于0wt%至1wt%的少量存在于组合物中。
[0068]
可以将抗氧化剂添加到组合物中以防止或最小化组合物中有机材料的氧化。合适的抗氧化剂包括例如,基于苯酚的抗氧化剂、由有机酸衍生物构成的抗氧化剂、含硫抗氧化剂、基于磷的抗氧化剂、基于胺的抗氧化剂、由胺-醛缩合物构成的抗氧化剂和由胺-酮缩合物构成的抗氧化剂。基于苯酚的抗氧化剂的实例包括经取代的苯酚,诸如1-氧基-3-甲基-4-异丙基苯、2,6-二-叔丁基苯酚、2,6-二-叔丁基-4-乙基苯酚、2,6-二-叔丁基-4-甲基苯
酚、4-羟基甲基-2,6-二-叔丁基苯酚、丁基羟基苯甲醚、2-(1-甲基环己基)-4,6-二甲基苯酚、2,4-二甲基-6-叔丁基苯酚、2-甲基-4,6-二壬基苯酚、2,6-二-叔丁基-α-二甲基氨基-对甲酚、6-(4-羟基-3,5-二-叔丁基苯胺基)2,4-双辛基-硫代-1,3,5-三嗪、正十八烷基-3-(4
′‑
羟基-3
′
,5
′‑
二-叔丁基苯基)丙酸酯、辛基化苯酚、经芳烷基取代的苯酚、烷基化对甲酚和受阻酚;双酚、三酚和多酚,诸如4,4
′‑
二羟基二苯基、亚甲基双(二甲基-4,6-苯酚)、2,2
′‑
亚甲基-双-(4-甲基-6-叔丁基苯酚)、2,2
′‑
亚甲基-双-(4-甲基-6-环己基苯酚)、2,2
′‑
亚甲基-双-(4-乙基-6-叔丁基苯酚)、4,4
′‑
亚甲基-双-(2,6-二-叔丁基苯酚)、2,2
′‑
亚甲基-双-(6-α-甲基-苄基-对甲酚)、亚甲基交联的多价烷基苯酚、4,4
′‑
亚丁基双-(3-甲基-6-叔丁基苯酚)、1,1-双-(4-羟基苯基)-环己烷、2,2
′‑
二羟基-3,3
′‑
二-(α-甲基环己基)-5,5
′‑
二甲基二苯基甲烷、烷基化双酚、受阻双酚、1,3,5-三甲基-2,4,6-三(3,5-二-叔丁基-4-羟基苄基)苯、三-(2-甲基-4-羟基-5-叔丁基苯基)丁烷、和四-[亚甲基-3-(3
′
,5
′‑
二-叔丁基-4
′‑
羟基苯基)丙酸酯]甲烷。合适的抗氧化剂是可商购的,例如,irganox
tm
抗氧化剂(汽巴特种化学品公司(ciba specialty chemicals corp.))。如果使用的话,抗氧化剂可以基于光致抗蚀剂底层组合物的总固体的例如大于0wt%至1wt%的量存在于组合物中。
[0069]
本发明的另一方面提供了一种经涂覆的基底,其包括布置在基底上的光致抗蚀剂底层组合物的层;以及布置在所述光致抗蚀剂底层组合物的层上的光致抗蚀剂层。所述经涂覆的基底可以进一步包括布置在光致抗蚀剂底层组合物之上和光致抗蚀剂层之下的含硅的层和/或有机减反射涂层。
[0070]
本发明的还另一方面提供了一种形成图案的方法。所述方法包括:(a)在基底上施加光致抗蚀剂底层组合物的层;(b)将所施加的光致抗蚀剂底层组合物固化以形成光致抗蚀剂底层;以及(c)在所述光致抗蚀剂底层上形成光致抗蚀剂层。所述方法可以进一步包括在形成所述光致抗蚀剂层之前,在所述光致抗蚀剂底层上形成含硅的层和/或有机减反射涂层。所述方法可以进一步包括图案化所述光致抗蚀剂层,并将所述图案从所述图案化的光致抗蚀剂层转移至所述光致抗蚀剂底层以及所述光致抗蚀剂底层下方的层。
[0071]
在所述图案化方法中可以使用各种各样的基底,其中电子装置基底是典型的。合适的基底包括例如,封装基底诸如多芯片模块;平板显示器基底;集成电路基底;用于包括有机发光二极管(oled)的发光二极管(led)的基底;半导体晶片;多晶硅基底;等。合适的基底可以呈晶片的形式,诸如用于制造集成电路、光学传感器、平板显示器、集成光学电路、和led的那些。如本文所用,术语“半导体晶片”旨在涵盖“电子装置基底”、“半导体基底”、“半导体装置”以及用于各种互连水平的各种封装物,包括单芯片晶片、多芯片晶片、用于各种水平的封装物、或其他需要焊接连接的组件。此类基底可以是任何合适的尺寸。典型的晶片基底直径是200mm至300mm,尽管根据本发明可以适当地使用具有更小和更大直径的晶片。如本文所用,术语“半导体基底”包括具有一个或多个半导体层或结构的任何基底,所述半导体层或结构可以任选地包括半导体装置的活性或可操作部分。半导体装置是指半导体基底,在其上已经批量制造或正在批量制造至少一种微电子装置。
[0072]
基底典型地由硅、多晶硅、氧化硅、氮化硅、氮氧化硅、锗化硅、砷化镓、铝、蓝宝石、钨、钛、钛-钨、镍、铜和金中的一种或多种构成。基底可以包括一个或多个层以及图案化特征。所述层可以包括例如一个或多个导电层,诸如铝、铜、钼、钽、钛、钨,此类金属的合金、氮
化物或硅化物、掺杂非晶硅或掺杂多晶硅的层;一个或多个介电层,诸如氧化硅、氮化硅、氮氧化硅或金属氧化物的层;半导体层,诸如单晶硅;以及其组合。所述层可以通过各种技术形成,例如化学气相沉积(cvd),诸如等离子体增强的cvd(pecvd)、低压cvd(lpcvd)或外延生长,物理气相沉积(pvd),诸如溅射或蒸发、或电镀。
[0073]
可以通过任何合适的手段诸如旋涂、狭缝式模头涂覆、刮涂、幕涂、辊涂、喷涂、浸涂等将光致抗蚀剂底层组合物涂覆在基底上。在半导体晶片的情况下,旋涂是优选的。在典型的旋涂方法中,将光致抗蚀剂底层组合物施加到以500至4000rpm的速率旋转的基底上持续15至90秒的时间段以在基底上获得希望的光致抗蚀剂底层组合物的层。本领域技术人员将理解,经涂覆的光致抗蚀剂底层组合物的厚度可以通过改变旋转速度以及光致抗蚀剂组合物的总固体含量来调节。由光致抗蚀剂底层组合物形成的光致抗蚀剂底层典型地具有5nm至50μm、典型地25nm至3μm、并且更典型地50至500nm的干燥层厚度。可以施加光致抗蚀剂底层组合物以基本上填充、优选填充、并且更优选完全填充基底上的多个间隙。
[0074]
任选地在相对较低的温度下将施加的光致抗蚀剂底层组合物软烘烤,以从所述组合物中去除任何溶剂和其他相对易挥发的组分。示例性的烘烤温度可以是60℃到170℃,尽管可以使用其他合适的温度。这种去除残余溶剂的烘烤可以进行10秒至10分钟,尽管可以适当地使用更长或更短的时间。当基底是晶片时,这种烘烤步骤可以通过在热板上加热所述晶片来进行。
[0075]
然后固化所施加的光致抗蚀剂底层组合物以形成光致抗蚀剂底层。光致抗蚀剂底层组合物应充分固化,使得所述光致抗蚀剂底层不与随后施加的层(诸如直接布置在光致抗蚀剂底层上的光致抗蚀剂层或其他有机层或无机层)混杂,或最小程度地与其混杂。可以在含氧气氛(诸如空气)中或在惰性气氛(诸如氮气)中并且在足以提供固化涂层的条件(诸如加热)下固化光致抗蚀剂底层组合物。此固化步骤优选在热板式设备上进行,尽管可以使用烘箱固化来获得等效的结果。固化温度应足以在整个层中进行固化,例如,足以使固化剂诸如游离酸进行交联,或使热酸产生剂释放酸并使释放的酸进行交联,其中所述固化剂是tag。典型地,固化在150℃或更高、并且优选150℃至450℃的温度下进行。更优选的是,固化温度是180℃或更高、还更优选200℃或更高、并且甚至更优选200℃至400℃。固化时间典型地是10秒至10分钟、优选30秒至5分钟、更优选45秒至5分钟、并且还更优选45至90秒。任选地,可以使用斜升式或多阶段固化工艺。斜升式烘烤典型地在相对低的(例如,环境)温度下开始,所述温度以恒定或变化的斜升速率增加至较高的目标温度。多阶段固化工艺涉及在两个或更多个温度平台处固化,典型地在较低的烘烤温度下进行第一阶段,在较高的温度下进行一个或多个额外的阶段。此类斜升式或多阶段固化工艺的条件对于本领域技术人员是已知的,并且可以允许省略先前的软烘烤工艺。
[0076]
在固化光致抗蚀剂底层组合物之后,可以将一个或多个处理层(诸如光致抗蚀剂层)、硬掩膜层(诸如金属硬掩模层)、有机或无机barc层等布置在固化的光致抗蚀剂底层上。光致抗蚀剂层可以直接在光致抗蚀剂底层的表面上形成,或者可替代地,可以在一个或多个中间层上的光致抗蚀剂底层上形成。在这种情况下,诸如以上所述的一个或多个中间处理层可以顺序地在光致抗蚀剂底层上形成,接着形成光致抗蚀剂层。合适的层、厚度和涂覆方法的确定是本领域技术人员熟知的。
[0077]
各种各样的光致抗蚀剂可以适当地用于本发明的方法中,并且典型地是正性材
料。合适的光致抗蚀剂包括例如从杜邦电子器件和成像公司(dupont electronics&imaging)(马萨诸塞州马尔堡(marlborough,massachusetts))可获得的epic
tm
系列光致抗蚀剂内的材料。可以通过已知的涂覆技术(诸如以上关于底层组合物描述的,其中旋涂是典型的)将光致抗蚀剂施加到基底上。光致抗蚀剂层的典型厚度是500至接下来,典型地将光致抗蚀剂层软烘烤以最小化所述层中的溶剂含量,从而形成无粘性涂层并提高所述层对基底的粘附性。软烘烤可以在加热板上或在烘箱中进行,其中加热板是典型的。典型的软烘烤在90℃至150℃的温度下进行,并且时间为30至90秒。
[0078]
任选地,可以在光致抗蚀剂层上布置一个或多个阻挡层。合适的阻挡层包括顶涂层、顶减反射剂涂层(或tarc层)等。优选地,当使用浸没式光刻将光致抗蚀剂图案化时,使用顶涂层。此类顶涂物在本领域中是熟知的,并且通常是可商购的,诸如可从杜邦电子器件和成像公司获得的oc
tm
2000。本领域技术人员将认识到,当在光致抗蚀剂层下面使用有机减反射剂层时,不需要tarc层。
[0079]
接下来,将光致抗蚀剂层通过光掩模暴露于活化辐射,以在曝光区域与未曝光区域之间产生溶解度差异。本文提及的将光致抗蚀剂组合物暴露于对所述组合物有活化作用的辐射表明辐射能够在所述光致抗蚀剂组合物中形成潜像。光掩模具有光学透明和光学不透明区域,分别对应于抗蚀剂层中的待通过活化辐射曝光的和未曝光的区域。曝光波长典型地是400nm以下,300nm以下,诸如248nm(krf)、193nm(arf)或euv波长(例如,13.5nm)。在优选的方面,曝光波长是193nm。曝光能量典型地是10至80mj/cm2,这取决于例如曝光工具和光敏组合物的组分。
[0080]
在暴露光致抗蚀剂层之后,典型地进行暴露后烘烤(peb)。peb可以例如在加热板上或在烘箱中进行。peb典型地在80℃至150℃的温度下进行,并且时间为30至90秒。由此形成由极性转换和未转换区域(分别对应于曝光和未曝光区域)之间的边界限定的潜像。然后使用适当的显影剂对曝光的光致抗蚀剂层进行显影,以提供图案化的光致抗蚀剂层。
[0081]
然后,可以通过适当的蚀刻技术(诸如通过等离子体蚀刻或湿蚀刻)将光致抗蚀剂层的图案转移到一个或多个包括光致抗蚀剂底层的下层并转移到基底。等离子体蚀刻可以对每个蚀刻的层使用适当的气体种类。合适的湿化学蚀刻化学法包括例如包含氢氧化铵、过氧化氢和水的混合物(例如,sc-1清洗液);包含盐酸、过氧化氢和水的混合物(例如,sc-2清洗液);包含硫酸、过氧化氢和水的混合物(例如,spm清洗液);包含磷酸、过氧化氢和水的混合物;包含氢氟酸和水的混合物;包含氢氟酸、磷酸和水的混合物;包含氢氟酸、硝酸和水的混合物;包含四甲基氢氧化铵和水的混合物;等。
[0082]
取决于所涉及的层和材料的数量,图案转移可以涉及使用不同技术的多个蚀刻步骤。在使用常规技术对基底进行图案转移之后,可以去除光刻堆叠件中的图案化的光致抗蚀剂层、光致抗蚀剂底层和其他任选的层。任选地,可以在图案转移到下层之后并且在图案转移到基底之前去除或消耗掉堆叠件的层中的一个或多个。然后根据已知方法进一步加工基底以形成电子装置。
[0083]
光致抗蚀剂底层组合物还可以用于自对准双图案化工艺。在这种工艺中,诸如通过旋涂将以上所述的光致抗蚀剂底层组合物的层涂覆在基底上。去除任何剩余的有机溶剂并且将涂层固化以形成光致抗蚀剂底层。将合适的中间层诸如含硅的硬掩膜层任选涂覆在光致抗蚀剂底层上。然后如通过旋涂将合适的光致抗蚀剂层涂覆在中间层上。然后对光致
抗蚀剂层进行成像(暴露),并且然后使用适当的显影剂对曝光的光致抗蚀剂层进行显影,以提供图案化的光致抗蚀剂层。接下来,通过适当的蚀刻技术将图案从光致抗蚀剂层转移到中间层以及光致抗蚀剂底层,以暴露基底的一部分。典型地,在此蚀刻步骤期间还将光致抗蚀剂去除。接下来,将共形含硅层置于图案化的光致抗蚀剂底层和基底的暴露部分之上。此类含硅层典型地是常规地通过cvd沉积的无机硅层,诸如sion或sio2。此类共形涂层产生在基底表面的暴露部分上以及在光致抗蚀剂底层图案之上的含硅层,即此类含硅层基本上覆盖了光致抗蚀剂底层图案的侧面和顶部。接下来,将含硅层部分地蚀刻(修整)以使图案化的光致抗蚀剂底层的顶表面和基底的一部分暴露。在此部分蚀刻步骤之后,基底上的图案包含多个特征,每个特征包含光致抗蚀剂底层的线或柱,其中含硅层直接与每个光致抗蚀剂底层特征的侧边相邻。接下来,诸如通过蚀刻去除光致抗蚀剂底层的暴露区域,以使在光致抗蚀剂底层图案下方的基底表面暴露,并且在基底表面上提供图案化的含硅层,其中与原始图案化的光致抗蚀剂底层相比,此类图案化的含硅层是双倍的(即,两倍多的线和/或柱)。
[0084]
由本发明的光致抗蚀剂底层组合物形成的光致抗蚀剂底层显示优异的平坦化、良好的耐溶剂性、和可微调的蚀刻速率。本发明的优选光致抗蚀剂底层组合物因此可用于各种半导体制造工艺。
[0085]
通过以下实例对本发明构思作进一步说明。本文使用的所有化合物和试剂都可商购获得,除了以下提供的程序。
[0086]
实例
[0087]
聚合物合成
[0088]
合成实例1
[0089]
向圆底烧瓶中添加10.0g的咔唑(1.5当量)、3.67g的乙醛酸一水合物(1当量)、和50ml的丙二醇单甲醚乙酸酯(pgmea)。将反应混合物温热至60℃并搅拌5分钟(min),接着一次性添加0.30g的甲磺酸。然后将反应物加热至120℃,持续16小时(h)。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤并用甲醇洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例1(p-1)。(66%产率,mw=2220,pdi=1.9)。
[0090]
合成实例2
[0091]
向圆底烧瓶中添加5.0g的咔唑(1.5当量)、1.85g的乙醛酸一水合物(1当量)、6.65g的1-丁醇(3当量)、和15ml的1,4-二噁烷。将反应混合物温热至60℃并搅拌5min,接着一次性添加1.45g的甲磺酸。然后将反应物加热至100℃,持续16h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并用甲醇洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例2(p-2)。(87%产率,mw=1510,pdi=1.4)。
[0092]
合成实例3至6
[0093]
用与以上相同的程序并用相应的醇(p-3:辛醇;p-4:2-(2-甲氧基乙氧基)乙醇;p-5:苄基醇;p-6:3,7-二甲基-1-辛醇)制备合成实例3(p-3)至6(p-6)以得到所需聚合物。
[0094][0095]
合成实例7
[0096]
向圆底烧瓶中添加6.0g的咔唑(1.25当量)、2.53g的丙酮酸(1当量)、和25ml的pgmea。将反应混合物温热至60℃并搅拌5min,接着一次性添加1.70g的甲磺酸。然后将反应物加热至120℃,持续16h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例7(p-7)。(76%产率,mw=3080,pdi=1.7)。
[0097]
合成实例8
[0098]
向圆底烧瓶中添加6.0g的咔唑(1.25当量)、3.33g的丙酮酸乙酯(1当量)、和25ml的pgmea。将反应混合物温热至60℃并搅拌5min,接着一次性添加1.70g的甲磺酸。然后将反应物加热至120℃,持续16h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例8(p-8)。(67%产率,mw=2630,pdi=1.5)。
[0099]
合成实例9
[0100]
向圆底烧瓶中添加5.0g的咔唑(1.5当量)、3.11g的三氟甲基丙酮酸甲酯(1当量)、和20ml的pgmea。将反应混合物温热至60℃并搅拌5min,接着一次性添加1.50g的甲磺酸。然后将反应物加热至120℃,持续16h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例9(p-9)。(65%产率,mw=10200,pdi=2.9)。
[0101][0102]
合成实例10
[0103]
向圆底烧瓶中添加6.0g的咔唑(1.5当量)、3.59g的苯基乙醛酸(1当量)、和25ml的pgmea。将反应混合物温热至60℃并搅拌5min,接着一次性添加1.70g的甲磺酸。然后将反应物加热至120℃,持续16h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例10(p-10)。(40%产率,mw=1250,pdi=1.2)。
[0104]
合成实例11
[0105]
向圆底烧瓶中添加10.0g的1-萘酚(1.2当量)、5.32g的乙醛酸一水合物(1当量)、和40ml的pgmea。将反应混合物温热至60℃并搅拌5min,接着一次性添加3.35g的甲磺酸。然后将反应物加热至120℃,持续20h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例11(p-11)。(88%产率,mw=3640,pdi=2.0)。
[0106]
合成实例12
[0107]
向圆底烧瓶中添加10.0g的1-萘酚(1.2当量)、5.32g的乙醛酸一水合物(1当量)、15.5g的1-丁醇(3当量)、和25ml的1,4-二噁烷。将反应混合物温热至60℃并搅拌5min,接着一次性添加3.35g的甲磺酸。然后将反应物加热至100℃,持续16h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例12(p-12)。(59%产率,mw=1750,pdi=1.4)。
[0108][0109]
合成实例13
[0110]
向圆底烧瓶中添加6.0g的1-萘酚(1.5当量)、2.45g的丙酮酸(1当量)、和25ml的pgmea。将反应混合物温热至60℃并搅拌5min,接着一次性添加2.00g的甲磺酸。然后将反应物加热至120℃,持续20h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例13(p-13)。(59%产率,mw=810,pdi=1.5)。
[0111]
合成实例14
[0112]
向圆底烧瓶中添加6.0g的1-萘酚(1.5当量)、3.22g的丙酮酸乙酯(1当量)、和25ml的pgmea。将反应混合物温热至60℃并搅拌5min,接着一次性添加2.00g的甲磺酸。然后将反应物加热至120℃,持续16h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例14(p-14)。(70%产率,mw=1010,pdi=1.7)。
[0113]
合成实例15
[0114]
向圆底烧瓶中添加5.0g的1-芘醇(1.5当量)、1.40g的乙醛酸一水合物(1当量)、和20ml的pgmea。将反应混合物温热至60℃并搅拌5min,接着一次性添加1.30g的甲磺酸。然后将反应物加热至120℃,持续20h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到合成实例15(p-15)。(62%产率,mw=1790,pdi=1.6)。
[0115][0116]
对比聚合物合成
[0117]
对比实例合成1
[0118]
向圆底烧瓶中添加5.0g的咔唑(1.5当量)、0.60g的多聚甲醛(1当量)、和20ml的pgmea。将反应混合物温热至60℃并搅拌5min,接着一次性添加1.45g的甲磺酸。然后将反应物加热至120℃,持续16h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v/)
的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到对比合成实例1(cp-1)。(80%产率,mw=2730,pdi=2.5)。
[0119]
对比实例合成2
[0120]
向圆底烧瓶中添加5.0g的咔唑(1.5当量)、1.85g的乙醛酸一水合物(1当量)、和25ml的1,4-二噁烷。将反应混合物温热至60℃并搅拌5min,接着一次性添加1.45g的甲磺酸。然后将反应物加热至100℃,持续16h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到对比合成实例2(cp-2)。(58%产率,mw=1610,pdi=1.3)。
[0121]
对比实例合成3
[0122]
向圆底烧瓶中添加5.0g的1-萘酚(1当量)、1.04g的多聚甲醛(1当量)、和25ml的pgmea。将反应混合物温热至60℃并搅拌5min,接着一次性添加0.20g的甲磺酸。然后将反应物加热至120℃,持续16h。在此反应时间之后,将反应混合物冷却至室温并倒入9/1(v/v)的甲醇/水中以得到固体聚合物产物。将产物过滤掉并在过量水和甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到对比合成实例3(cp-3)。(52%产率,mw=2240,pdi=2.0)。
[0123]
对比实例合成4
[0124]
向圆底烧瓶中添加5.0g的1-芘醇(1当量)、0.69g的多聚甲醛(1当量)、和20ml的丙二醇单甲醚(pgme)。将反应混合物温热至60℃并搅拌5min,接着一次性添加2.20g的甲磺酸。然后将反应物加热至120℃,持续1h。在此反应时间之后,反应物大部分固化。将固体从烧瓶中取出并在过量水和甲醇中洗涤,然后空气干燥4h并在50℃下真空干燥,持续额外的20h,以得到对比合成实例4(cp-4)。
[0125][0126]
物理测试
[0127]
聚合物的数均分子量和重均分子量(分别是mn和mw)以及多分散性(pdi)值(mw/mn)通过在配备有agilent 1100系列折射率和minidawn光散射检测器(怀雅特技术公司(wyatt technology co.))的agilent 1100系列lc系统上进行凝胶渗透色谱法(gpc)来测量。将样品以大约10mg/ml的浓度溶解于hplc等级thf中,并且通过0.45μm注射器式过滤器过滤,然后注入通过四个shodex柱(kf805、kf804、kf803和kf802)。维持1ml/min的流速和35℃的温度。用窄分子量ps标准物(easical ps-2,聚合物实验室有限公司(polymer laboratories,inc.))校准所述柱。
[0128]
使用差示扫描量热法(dsc)来确定本体聚合物上的玻璃化转变温度。将样品(1-3mg)加热至150℃并在150℃下静置10min以去除第一个循环上的残余溶剂,然后冷却至0℃
并以10℃/min的加热速率斜升至300℃。使用第二加热曲线和可逆加热曲线来鉴定玻璃化转变温度。
[0129]
表1示出了聚合物p-1至p-10和对比聚合物cp-1和cp-2的分子量、溶解度、和热表征。
[0130]
表1.
[0131]
实例mwpdi溶解度atg(℃)bp-122201.9 164p-215101.4 108p-319301.4 67p-417601.5 114p-515401.4 84p-619801.4 29p-730801.7 166p-823601.5 157p-9102002.9 n/ap-1012501.2 n/acp-127302.5xn/acp-216101.3xn/a
[0132]
a.在pgmea中在10wt%下测量的溶解度。 指示完全溶解,x指示在此浓度下微小溶解或不溶解。
[0133]
b.通过dsc测量,加热速率10℃/min;n/a指示“未观察到”。
[0134]
表2示出了聚合物p-11至p-15和对比聚合物cp-3和cp-4的分子量、溶解度、和热表征。
[0135]
表2.
[0136]
实例mwpdi溶解度atg(℃)bp-1136402.0 177p-1217501.4 156p-138101.5 117p-1410101.7 141p-1517901.6 186cp-322402.0 n/acp-4
‑‑
xn/a
[0137]
a.在pgmea中在10wt%下测量的溶解度。 指示完全溶解,x指示在此浓度下微小溶解或不溶解。
[0138]
b.通过dsc测量,加热速率10℃/min;n/a指示“未观察到”。
[0139]
如可以从表1和表2中所见,与对比实例相比,本发明的组合物具有更好的溶解度和更低的玻璃化转变温度。
[0140]
配制品
[0141]
通过将表1和表2中所列的聚合物与表3中概述的组分合并以形成光致抗蚀剂底层
wave co.)的optiprobe
tm
测量膜厚度。将丙二醇单甲醚乙酸酯(pgmea)施加到膜上,持续90s,接着在105℃下进行剥离后烘烤(psb),持续60s。根据等式1计算耐溶剂性:
[0150]
[(剥离前的ft)-(psb后的ft)]/(剥离前的ft)*100%等式1
[0151]
其中ft是膜厚度。耐溶剂性在表5中报告,其中a被定义为99%-100%耐溶剂性,并且b被定义为90%-99%耐溶剂性。
[0152]
蚀刻速率
[0153]
针对以100-200nm的膜厚度在8英寸硅晶片上涂覆并烘烤的光致抗蚀剂底层组合物确定蚀刻速率。使用plasma-therm 700 系列蚀刻工具来使用表4中所示的条件确定主体膜干蚀刻速率。使用o2或cf4等离子体蚀刻固化的光致抗蚀剂底层组合物。作为时间的函数测量蚀刻之前和之后的膜厚度,并且计算蚀刻速率。光致抗蚀剂底层组合物的蚀刻速率在表5中示出。
[0154]
表4.
[0155]
前体cf4ar/o2流量(sccm)5060/20功率(w)500300压力(mt)1010时间(s)30,60,12030,60,90
[0156]
平坦化测试
[0157]
评估本发明的光致抗蚀剂底层组合物来确定它们的平坦化特性。在cnse nano-fab(纽约奥尔巴尼(albany,ny))创建模板。所述模板具有100nm的sio2膜厚度、以及各种间距和图案,模头大小为1cm x 1cm。每个模头以100nm间隔的台阶图案开始,接着是2000μm的非图案开放区域,接着是覆盖45nm/90nm至2μm/5μm间距沟槽的各种线/空间图案。使用第一台阶图案来判断平坦化性能。在用本发明的组合物涂覆试样之前,将模板试样在150℃下烘烤60秒作为脱水烘烤。使用旋涂机和1500rpm /-200rpm的旋转速率将每个光致抗蚀剂底层组合物涂覆在模板试样上。固化之后的目标膜厚度是100nm,并且相应地调节组合物稀释度以近似地得到固化之后的目标膜厚度。通过在240℃下将晶片放置在热板上60秒来将膜固化。通过kla tencor p-7stylus轮廓仪评估跨台阶的膜的平坦化质量。
[0158]
在表5中,平坦化质量如下定义:a指示小于20nm的高度变化,b指示在20-28nm之间的高度变化,并且c指示大于28nm的高度变化。更低的数值指示优异的平坦化性能,所以a表示最佳平坦化,接着是b,并且c表示最差的平坦化性能。
[0159]
表5.
[0160][0161]
如可以从表5中所见,本发明的光致抗蚀剂底层组合物具有优异的平坦化性能,并且具有可以通过共混或混合树脂来微调至高于、低于或匹配对比组合物的蚀刻速率。
[0162]
虽然已经结合目前被认为是实际的示例性实施例描述了本公开,但是应当理解,本发明不限于所公开的实施例,而且相反地,旨在覆盖包括在所附权利要求的精神和范围内的各种修改和等同布置。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。