使用带电表面从诱导多能干细胞产生多个谱系的方法
1.本技术要求于2019年6月14日提交的美国临时专利申请第62/861,640号、2019年6月24日提交的第62/865,806号和2020年6月12日提交的第63/038,564号的权益,其全部内容通过引用并入本文。
2.背景
3.1.领域
4.本发明一般涉及分子生物学和医学领域。更具体地说,它涉及分化诱导多能干细胞的方法。
5.2.相关技术说明
6.人多能干细胞(hpsc)为再生医学和药物开发中的应用提供了强大的资源。在过去的十年中,已经开发出各种方法来使用多种生长成分、细胞外基质和/或饲养层从ipsc生成多个谱系,从而为改进体外模型和研究不同谱系的早期发育提供潜在的强大工具。
7.有效体外分化成特定细胞类型是其用于疾病建模和药物筛选的潜在用途的关键。谱系特异性分化的关键成功因素包括使用确定的条件、终末期细胞的良好表型和功能表征、分化的过程长度的可重复性和成本。因此,本领域仍然需要用于人多能干细胞谱系的特异性分化的有效方法。
8.概述
9.本公开内容的某些实施方案提供用于分化诱导多能干细胞(ipsc)的体外方法的方法和组合物,包括:(a)在不存在细胞外基质蛋白的情况下在带电表面上培养ipsc;和(b)将ipsc分化为内皮细胞、间充质干细胞(msc)或造血前体细胞(hpc)。
10.在一些方面,带电表面带正电。在某些方面,带正电的表面是胺表面或聚l赖氨酸表面。在特定方面,带正电的表面包含含氮官能团。在其他方面,带电表面带负电。在特定方面,带负电的表面是羧基表面。在某些方面,带负电的表面包含含氧官能团。在一些方面,带电表面是聚合物表面。例如,聚合物表面是聚苯乙烯表面。在某些方面,带电表面包含带正电的基团和带负电的基团。在一些方面,带正电的基团是含氮基团,并且带负电的基团是含氧基团。
11.在某些方面,ipsc在无血清的成分确定培养基中培养。在一些方面,分化包括在blebbistatin或rock抑制剂如h1152的存在下培养。在特定方面,该方法不含或基本上不含细胞外基质蛋白,例如层粘连蛋白、纤连蛋白、玻连蛋白、matrigel
tm
、腱生蛋白、巢蛋白、血小板反应蛋白、弹性蛋白、明胶或胶原蛋白。
12.在另外的方面,该方法进一步包括在步骤(a)之前工程化ipsc以具有破坏的trem2、mecp2和/或scna的表达。在特定方面,工程化包括使用tal核酸酶在trem2的外显子2中引入插入缺失。在一些方面,破坏的mecp2的表达被进一步定义为mecp2蛋白的截短突变体。在某些方面,破坏的表达是由于错义点突变例如a53t造成的。
13.在一些方面,该方法包括将祖细胞分化为内皮细胞。在某些方面,步骤(a)包括在胺表面上培养以产生祖细胞并且步骤(b)包括在内皮分化培养基的存在下在羧基表面上培养以产生内皮细胞。在特定方面,内皮细胞对cd31呈阳性。
14.在进一步的方面,该方法进一步包括将内皮细胞分化为脑微血管内皮细胞(bmec)。
15.在一些方面,该方法进一步包括将内皮细胞分化为淋巴管内皮细胞。
16.在某些方面,该方法包括将祖细胞分化为msc。在特定方面,分化包括在存在msc培养基的情况下在胺表面上培养祖细胞。在某些方面,msc对cd73、cd44和cd105呈阳性。在某些方面,至少90%的分化细胞对cd73呈阳性。
17.在另外的方面,该方法进一步包括将msc分化为周细胞。在某些方面,在不存在胞外蛋白的情况下在周细胞培养基的存在下培养msc。在一些方面,周细胞对ng2、pdgfrβ和cd146呈阳性。
18.在一些方面,该方法包括将祖细胞分化为hpc。在某些方面,该方法还包括将hpc分化为小胶质细胞。在某些方面,分化包括在存在小胶质细胞分化培养基的情况下在中性带电的表面或超低附着表面上培养hpc。在某些方面,小胶质细胞分化培养基包含il34、tgf和mcsf。在一些方面,分化包括在常氧下培养。在一些方面,分化持续20-25天。在特定方面,小胶质细胞对cd45、cd11b和cd33呈阳性。在某些方面,至少50%(例如,51%、52%、53%、54%、55%、56%、57%、58%、59%、60%、65%、70%、75%、80%、85%、90%、50-60%、60-70%或80-90%)的分化细胞对cd11b呈阳性。在一些方面,至少90%(例如,91%、92%、93%、94%、95%、96%、97%、98%、99%或100%)的分化细胞对cd33呈阳性。
19.在某些方面,该方法不包括细胞的纯化。在一些方面,纯化进一步定义为进行macs。
20.在特定方面,该方法符合良好制造规范(gmp)。在一些方面,该方法在缺氧条件下进行。在具体方面,ipsc是人的。
21.在另一个实施方案中,提供了一种包含小胶质细胞群的组合物,其中至少90%(例如,91%、92%、93%、94%、95%、96%、97%、98%、99%、100%、90-93%、93-96%或96-100%)的小胶质细胞群对p2ry12、cx3cr1、tmem119、iba-1和trem2呈阳性。在一些方面,小胶质细胞群是通过实施方案的方法产生的。在一些方面,小胶质细胞群从具有trem2、apoe、cd33、bin、abca7、snps或与神经变性相关的基因型的疾病相关ipsc供体产生。在某些方面,小胶质细胞群具有破坏的trem2、mecp2和/或scna的表达。在一些方面,破坏的trem2的表达进一步定义为trem2表达的纯合敲除。在某些方面,破坏的mecp2的表达进一步定义为mecp2蛋白的截短突变体。在一些方面,破坏的scna的表达是由于错义点突变例如a53t造成的。
22.另一个实施方案提供了筛选测试化合物的方法,包括将测试化合物引入本发明实施方案的小胶质细胞群。在一些方面,将小胶质细胞群进一步引入淀粉样蛋白-β。在某些方面,将小胶质细胞群进一步引入lps。
23.另一个实施方案提供了包含通过本实施方案的方法产生的周细胞细胞群的组合物。
24.在又一个实施方案中,本文提供了一种血脑屏障模型,其包含由本发明实施方案产生的小胶质细胞、周细胞和bmec。
25.另一个实施方案提供了一种产生小胶质细胞的方法,包括:(a)将ipsc分化为hpc;(b)针对cd34阳性细胞分选hpc;和(c)在小胶质细胞分化培养基中培养hpc,从而产生小胶质细胞群。在一些方面,根据本发明实施方案分化hpc。在某些方面,分选包括使用cd34磁
珠。在特定方面,该方法不包括针对cd43阳性细胞分选hpc。在特定方面,该方法不包括ecm蛋白。
26.在一些方面,小胶质细胞分化培养基包含il-34、tgfβ1或m-csf。在某些方面,小胶质细胞分化培养基包含200ng/ml il-34、100ng/ml tgfβ1和50ng/ml m-csf。在一些方面,细胞每48小时用小胶质细胞分化培养基进行补料。
27.在特定方面,小胶质细胞群中至少90%(例如,91%、92%、93%、94%、95%、96%、97%、98%、99%、100%、90-93%、93-96%或96-100%)的细胞是trem阳性的。在一些方面,至少10%(例如,15%、20%、25%、30%、10-15%、15-20%或20-30%)的hpc分化为小胶质细胞。
28.在某些方面,步骤(b)的培养以96孔格式进行。在某些方面,步骤(b)的培养在带电表面上进行。在一些方面,带电表面带正电。例如,带正电的表面是胺表面。在其他方面,带电表面带负电。例如,带负电的表面是羧基表面。
29.在另外的方面,该方法进一步包括在包含cd200和/或分形趋化因子的培养基中使小胶质细胞群成熟。在一些方面,该方法进一步包括冷冻保存小胶质细胞。
30.在一些方面,hpc从被工程化以破坏trem2表达的ipsc分化而来。在某些方面,工程化包括使用tal核酸酶。
31.在某些方面,冷冻保存的小胶质细胞保留针对phrodo细菌颗粒的吞噬功能。在特定方面,冷冻保存的小胶质细胞能够在解冻后成熟,并对刺激物作出反应,并在培养基上清中分泌白介素、趋化因子和免疫调节配体。
32.另一个实施方案提供了一种用于从ipsc产生神经祖细胞(npc)的体外方法,包括(a)在包含糖原合酶激酶3(gsk3)抑制剂的培养基中预处理(pre-condition)ipsc;(b)将ipsc分化为npc,其中该方法不包括抑制smad信号传导。
33.在一些方面,在步骤(a)之前将ipsc维持在缺氧条件下。在某些方面,在步骤(a)之前,在rock抑制剂的存在下接种ipsc,然后在不存在rock抑制剂的情况下培养ipsc。
34.在某些方面,gsk3抑制剂是chir99021、bio或sb-216763。在特定方面,gsk3抑制剂是chir99021,例如浓度为1μm、2μm、3μm、4μm或5μm,特别是3μm。在一些方面,预处理持续2-4天,例如1、2或3天。
35.在一些方面,步骤(a)和/或步骤(b)的ipsc在细胞外基质(ecm)蛋白包被的表面上培养。在某些方面,ecm蛋白是matrigel
tm
、层粘连蛋白或玻连蛋白。在具体方面,ecm蛋白是层粘连蛋白或玻连蛋白。
36.在某些方面,步骤(a)和(b)在常氧条件下进行。在一些方面,分化包括在ecm蛋白包被的表面上培养ipsc。在某些方面,ecm蛋白是层粘连蛋白或玻连蛋白。在一些方面,分化包括在rock抑制剂的存在下在超低附着板或旋转瓶上培养ipsc。在特定方面,步骤(b)进行5至10天,例如6天、7天、8天、9天或10天。
37.在另外的方面,该方法还包括检测npc中tra-162、cd56、cd15、sox1、神经上皮干细胞蛋白、β3微球蛋白和/或pax-6的表达。在一些方面,至少70%(例如,80%、85%、90%、95%、70-80%、80-90%或90-100%)的npc对cd56呈阳性。
38.在进一步的方面,该方法包括将npc进一步分化为星形胶质细胞或神经元。
39.另一个实施方案提供了一种筛选神经变性疾病的方法,包括检测小胶质细胞条件
培养基中可溶性trem2的水平。在一些方面,检测包括进行elisa。在某些方面,小胶质细胞源自等基因工程化的ipsc或表达疾病相关snp或突变的供体。在一些方面,小胶质细胞通过本发明实施方案或其方面的方法产生。在某些方面,与对照相比增加的可溶性tem水平检测神经变性疾病,例如阿尔茨海默病或多发性硬化症。
40.另一个实施方案提供了一种用于进行高通量筛选以鉴定治疗剂的方法,包括使通过本发明实施方案或其方面的方法产生的小胶质细胞与多种候选剂接触并测量细胞因子和/或趋化因子水平和/或淀粉样蛋白β吞噬功能。
41.在一些方面,小胶质细胞是冷冻保存的源自等基因工程化的ipsc系的小胶质细胞、源自表达疾病相关snp的供体的小胶质细胞或源自表达与神经变性相关的突变的供体的小胶质细胞。在一些方面,细胞因子和/或趋化因子选自il6、il10、il3、tnfα、il13、ccl2/mcp-1、ccl20/mip-3α、ccl4/mip-1β、ccl5/rantes、cx3cl1/分形趋化因子、cxcl1/groα、cxcl10/ip-10、cxcl2/groβ和il-8/cxcl8。
42.本文还提供了包含本发明实施方案及其方面的小胶质细胞和内皮细胞、周细胞、星形胶质细胞和/或神经前体细胞的共培养物。另一个实施方案提供了该共培养物在模拟人脑发育中的用途。
43.本发明的其他目的、特征和优点将从以下详述中变得明显。然而,应当理解,详述和具体实施例虽然指示了本发明的优选实施方案,但仅以举例说明的方式给出,因为根据该详述,在本发明的精神和范围内的各种变化和修改对本领域技术人员来说将是明显的。
44.附图的简要说明
45.以下附图构成本说明书的一部分并且被包括在内以进一步说明本发明的某些方面。通过参考这些附图中的一幅或多幅并结合在本文中呈现的具体实施方案的详述,可以更好地理解本发明。
46.图1:2d和3d hpc分化过程的示意图。
47.图2:从源自2d或3d hpc分化过程的第6天hpc衍生出内皮细胞的示意图。
48.图3:在不使用cd31
macs的情况下使用连续传代纯化生成的内皮细胞的表征。
49.图4a-b:(图4a)重新铺板3结束时细胞的形态学。内皮细胞可以在重新铺板传代2或3结束时冷冻保存。(图4b)用于衍生内皮细胞的培养基配方。
50.图5a-5d:(图5a)msc分化过程的概述。(图5b)用于产生msc的培养基配方。(图5c)描述msc向脂肪细胞、骨细胞和软骨的三系分化的示意图。(图5d)第6天msc祖细胞的表型表征。
51.图6:在胺表面出现纯msc群。将冷冻保存的第6天hpc或第6天分化结束时的活培养物在msc培养基和1um h1152(或blebbistatin)的存在下置于胺带电表面平板上。在p4时将培养物转移至常氧和正常组织培养板。在p5时达到了msc的纯度规格。
52.图7:针对表面msc标志物cd73、cd44、cd105、cd49d以及内皮标志物cd31和cd144的缺失染色的细胞。将冷冻保存的第6天hpc或第6天分化结束时的活培养物在msc培养基和1um h1152(或blebbistatin)的存在下置于胺带电表面平板上。在p5时将培养物转移至常氧和正常组织培养板。在p6时达到了msc的纯度规格。
53.图8a-8c:(图8a)三系潜能。从msc生成脂肪细胞、骨细胞和软骨细胞的各个步骤。(图8b)展示msc的三系潜能、骨细胞的茜素红染色、软骨细胞的阿尔新蓝和脂肪细胞的油红
o的图片。(图8c)msc以1,000个细胞/cm2接种在msc分化培养基中并且每隔一天补料持续10-14天。使用结晶紫对平板染色并计算集落总数。
54.图9a-9h:msc向周细胞的转化。(图9a)将icell msc转化为ipsc衍生的周细胞的过程的示意图。(图9b)用于产生ipsc衍生的周细胞的培养基配方。(图9c)icell msc、ipsc衍生的周细胞和sciencell原代人脑血管周细胞(hbvp)中已知周细胞标志物pdgfrβ、ng2和cd146的比较流式细胞术。icell msc在解冻时没有周细胞标志物,而在周细胞培养基中p1结束时获得周细胞标志物。ipsc衍生的周细胞显示出比原代hbvp更高纯度的周细胞特异性标志物。(图9d)ipsc衍生的msc(p2)、ipsc衍生的周细胞(p1)和sciencell原代hbvp形态的通过明视野显微术的形态学。(图9e)描述基于表型和标志物表达的pc1和pc2周细胞亚型之间的差异的表。(图9f)ipsc衍生的周细胞在解冻后立即和在解冻后五天通过流式细胞术针对周细胞亚型特异性标志物cd274、vcam1、结蛋白、dlk1和αsma以及通用周细胞标志物pdgfrβ、ng2、cd13和cd146进行染色。ipsc衍生的周细胞揭示了收缩周细胞pc2亚型的特征。(图9g)在吞噬作用测定中ipsc衍生的周细胞的incucyte实时成像系统图像。ipsc衍生的周细胞显示出金黄色葡萄球菌生物颗粒的高于对照水平的可观察到的吞噬活性(a)ipsc衍生的周细胞单独对照。(b)ipsc衍生的周细胞 nucgreen dead 488(ng)试剂对照。(c)ipsc衍生的周细胞 金黄色葡萄球菌phrodo red生物颗粒(bp)。(d)ipsc衍生的周细胞 nucgreen dead 488试剂(ng) 金黄色葡萄球菌phrodo red生物颗粒(bp)。从细胞接种后36天16小时的时间点拍摄的所有图像。(图9h)通过incucyte软件分析的总红色物体综合强度定量吞噬活性。
55.图10a-10g:脑微血管内皮细胞(bmec)的产生。(图10a)产生脑微血管内皮细胞的示意图概述。(图10b)ecra培养基的组成。(图10c)通过glut1/cd31的共表达对bmec进行流式细胞术分析。(图10d)在第13天具有p糖蛋白(绿色)表达的bmec的免疫组织化学染色。用hoechst3342染色细胞核并且通过imagexpress(molecular devices,llc)以200倍放大率捕获图像。(图10e)通过测量铺板后数天的teer值对bmec进行功能表征。(图10f)使用替代方法生成脑微血管内皮细胞的示意图概述,该替代方法包括预处理步骤和在不使用ecm的情况下在带电表面上铺板。在带电表面上诱导bmec生成的改良培养基的描述。(图10g)收获在不同带电表面上的第7天分化脑微血管内皮细胞,并通过针对cd31、p糖蛋白/和glut-1表达的存在以及多能性标志物(tra-181)表达的缺乏的染色来定量纯度。
56.图11:使用3d分化过程按比例增加hpc,随后使用cd34
磁珠进行纯化。使用cd34磁珠按比例增加和分选hpc的示意图。举例说明了通过手动和clinimacs介导的分离进行分选过程的效率。概述了每次运行的hpc的实际纯度(通过分选的级分中cd34阳性细胞的百分比来测量)和该过程的效率。
57.图12:用于小胶质细胞分化的培养基配方。
58.图13a-13b:(图13a)从cd34 分选的hpc衍生小胶质细胞的示意图。将hpc铺板在小胶质细胞分化培养基mdm中。培养物每48小时用mdm或2x mdm进行补料。培养物在分化的第12天分开(split),并且2d分化一直持续到第23天。在第23天收获细胞,并针对小胶质细胞培养物的纯度标志物的存在进行染色。其余的培养物被冷冻保存。在冷冻保存之前和之后定量小胶质细胞培养物的纯度。(图13b)描绘了小胶质细胞分化培养基和小胶质细胞分化培养基。
59.图14:冷冻保存之前和之后在mdm培养基的存在下的小胶质细胞纯度评估。收获第23天分化时的小胶质细胞培养物,并针对小胶质细胞特异性标志物的存在进行染色。使用速率受控冷冻机冷冻保存剩余的细胞。将冷冻保存的细胞解冻并针对小胶质细胞特异性标志物的存在进行染色。对于这两组,通过流式细胞术评估cd45、cd33、trem2和cd11b的细胞表面表达(图14a)以及cx3cr1pu.1、iba、p2ry12、trem2和tmem119的细胞内表达。
60.图15a-c:使用手动相对速率受控冷冻机在冷冻保存后恢复小胶质细胞。将hpc置于培养基中以在mdm的存在下启动小胶质细胞分化。在分化的第20天(图15b)、第23天(图15b)和第26天(图15c)使用手动冷冻方案或速率受控冷冻机(crf)将细胞冷冻保存。将冷冻保存的细胞转移到液氮中一周。将冷冻保存的小胶质细胞解冻并置于小胶质细胞成熟培养基(mmm)中。培养物每48小时补料新鲜成熟培养基。在解冻后第3、5、7、10、12和14天收获细胞,并定量相对于初始铺板数的活细胞回收率。
61.图16:将hpc转化为小胶质细胞的效率。冷冻保存的hpc在mdm的存在下分化为小胶质细胞(n=4)。输入hpc和输出小胶质细胞的总存活数被定量。基于小胶质细胞分化的第23天存在的trem2阳性细胞的纯度和绝对数量除以输入活hpc的绝对数量来计算过程效率。
62.图17a-17c:在解冻的第20天(图17a)、第23天(图17b)和第26天(图17c)、解冻后3天和解冻后10天手动地或在速率受控冷冻机的存在下冷冻保存的小胶质细胞的纯度分析。将在第20、23和26天冷冻保存的小胶质细胞解冻并在小胶质细胞成熟培养基中铺板3、5、7、10和12天。通过流式细胞术针对pu1、iba、cx3cr和p2ry12表达的存在对细胞进行染色。
63.图18a-18b:在解冻后第0天(图18a)和第3天(图18b)手动或在速率受控冷冻机的存在下冷冻保存的小胶质细胞的手动血细胞计数器活细胞计数,以设置使用金黄色葡萄球菌生物颗粒的吞噬测定。
64.图19:使用手动或速率受控冷冻机在分化的第20、23和26天冷冻保存的小胶质细胞的功能表征。在每孔存在200μl小胶质细胞成熟培养基的情况下,将冷冻保存的小胶质细胞解冻并以15,000个活细胞/孔铺板在96孔板中。用稀释的1μg/孔的调理或非调理phrodo red生物颗粒(thermo fisher#a10010,每瓶2mg;储存在-20℃)处理细胞。将板置于incucyte上,并在解冻后最多5天的不同时间点拍摄吞噬作用的图像。通过速率受控冷冻方法冷冻保存的细胞表现出更强大的吞噬作用(由于更高的细胞活力)。手动冷冻保存方法在所有条件下显示吞噬作用的降低/右移的速率(由于细胞活力降低)。
65.图20:通过incucyte系统上的实时成像评估的活的第14天小胶质细胞和在分化的第20、23天和第26天使用手动或速率受控冷冻机冷冻保存的小胶质细胞的功能表征。将冷冻保存的小胶质细胞解冻并铺板在mmm中三天。三天结束时的活细胞计数如图18b中所述确定。在存在每孔200μl小胶质细胞成熟培养基(mmm)的情况下,将15,000个活细胞铺板到96孔板中。每48小时给细胞补料新鲜的50μl mmm培养基。用稀释的1μg/孔的调理或非调理phrodo red生物颗粒(thermo fisher#a10010,每瓶2mg;储存在-20℃)处理细胞。将板置于incucyte上,并在解冻后5、7和14天的不同时间点拍摄吞噬作用的图像。手动冷冻保存方法显示在所有条件下吞噬作用降低/右移的速率(由于细胞活力降低)。
66.图21:通过将来自解冻后样品的吞噬红细胞计数除以总细胞数确定吞噬效率比。
67.图22:使用phrodo淀粉样蛋白β对冷冻保存的小胶质细胞进行功能表征。在存在每孔200μl小胶质细胞成熟培养基的情况下,将冷冻保存的第23天的小胶质细胞解冻并以15,
000个活细胞/孔铺板在96孔板中。用phrodo淀粉样蛋白β处理细胞。对照组包含带有不含任何phrodo淀粉样蛋白β的培养基的细胞。将板放置在incucyte上,并在解冻后最多24小时的不同时间点拍摄吞噬作用的图像。
68.图23a-23b:在没有ecm的情况下,和在适合筛选应用的96孔格式中,将hpc的分化微型化至小胶质细胞。hpc分化为小胶质细胞的示意图(图23a)和实验中使用的不同带电表面。超低附着(ula)、组织培养(tc)和非组织培养(non-tc)容器(图23b)。
69.图24a-24b:在各种带电表面的存在下第23天小胶质细胞的末期纯度分析。将冷冻保存的hpc以20,000-35,000个活细胞/cm2的密度在每孔200μl小胶质细胞分化培养基的存在下铺板在96孔primaria板或超低附着、组织培养(tc)或非组织培养板(non-tc)上。在接下来的23天分化中,细胞每48小时用每孔mdm 50μl培养基进行补料。在第23天用冷pbs收获细胞,并使用自动细胞计数器定量总活细胞数。针对cd11b、cd45、cd33、trem2的表面表达和trem2、iba、p2ry12和tmem119的细胞内表达对细胞进行染色。
70.图25a-25b:冷冻保存的小胶质细胞释放的细胞因子和趋化因子。将第23天冷冻保存的小胶质细胞解冻到mdm培养基中并以50,000个细胞/孔铺板到primaria 96孔板上。在开始用100ng/ml lps和50ng/ml干扰素γ刺激之前,将细胞铺板三天。刺激一式三份进行,持续24小时。将上清液离心以去除细胞和碎片,并立即置于-20℃。在多重luminex测定上分析上清液。显示了多个wt批次(图25a)和wt、纯合和杂合trem2敲除(ko)、mecp2 hm和a53t-snca工程化系(图25b)的平均值。
71.图26:来自纯合和杂合trem2基因敲除(ko)、mecp2和a53t-snca工程化系的多个批次的冷冻保存的小胶质细胞针对cd11b、cd45、trem2的表面表达以及细胞内标志物pu.1、iba-1、cx3cr1、p2ry12和tmem119的存在进行染色。工程化的ipsc系通过流式细胞术显示了可比较的trem2表达。
72.图27a-27b:(图27a)在源自野生型(wt)、杂合(ht)和纯合(ho)trem2 ko工程化ipsc的冷冻保存的小胶质细胞中可溶性trem2的释放。使用simple step elisa(abcam)从来自wt和trem2杂合和纯合ko突变体的条件培养基中定量可溶性trem2(strem2)的水平。将wt和trem2 ko小胶质细胞解冻并在96孔primaria板中的成熟培养基中以相同的密度铺板。在解冻后第3天和解冻后第7天收集用过的培养基。在解冻后的第3天和第5天,培养物用新鲜成熟培养基进行半补料。(图27b)在源自野生型(wt)、mecp2hm和a53t-snca工程化ipsc的冷冻保存的小胶质细胞中可溶性trem2的释放。使用simple step elisa(abcam)从来自wt、mecp2hm和a53t-snca工程化系的条件培养基中定量可溶性trem2(strem2)的水平。将小胶质细胞解冻并以相同的密度铺板在96孔primaria板中的成熟培养基中。在解冻后第3天和解冻后第7天收集用过的培养基。在解冻后的第3天和第5天,培养物用新鲜成熟培养基进行半补料。
73.图28:用于研究wt、ht和ho trem2 ko工程化小胶质细胞wt、1185ht trem2 ko、1187ho trem2 ko的存活动力学的培养基配方的列表。将小胶质细胞以每孔15,000个活细胞的密度置于96孔板中的250μl含有32种不同细胞因子配方的小胶质细胞基础培养基中。在incucyte系统上捕获细胞存活的动力学。将稀释至2滴/ml的nucgreen dead加入基于不同培养基成分的含有细胞的所有孔中,以随时间捕获死细胞的数量。每8小时拍摄一次图像,实验在没有任何间歇性进料的情况下持续72小时。
74.图29a-29c:wt(图29a)、1185ht trem2 ko(图29b)、1187ho trem2 ko(图29c)小胶质细胞存活动力学。
75.图30a-30c:使用两种细胞因子的wt(图30a)、1185ht trem2 ko(图30b)、1187ho trem2 ko(图30c)小胶质细胞存活动力学。
76.图31a-31c:使用三种细胞因子的wt(图31a)、1185ht trem2 ko(图31b)、1187ho trem2 ko(图31c)小胶质细胞存活动力学。
77.图32a-32c:使用四种细胞因子的wt(图32a)、1185ht trem2 ko(图32b)、1187ho trem2 ko(图32c)小胶质细胞存活动力学。
78.图33a-33e:将wt、1185ht trem2 ko、1187ho trem2 ko小胶质细胞以15,000个活细胞的密度置于96孔板中的250μl小胶质细胞基础培养基(图33a)或mmm(图33b),或补充有il-34的小胶质细胞基础培养基(图32c),或补充有il-34的小胶质细胞基础培养基(图33d),或补充有mcsf的小胶质细胞基础培养基(图33d)或仅补充有il-34的基础培养基(图33c)或mscf或il-34和mcsf的组合(图33e)中。在incucyte系统上捕获细胞存活的动力学。将稀释至2滴/ml的nucgreen dead加入含有细胞的具有不同培养基成分的所有孔中,以随时间捕获死细胞的数量。每8小时拍摄一次图像,实验在没有任何间歇性进料的情况下继续7天。nucgreen dead的强度定量了培养物中的死细胞。
79.图34a-34h:在第23天冷冻保存的wt、1185 ht trem2 ko、1187 ho trem2 ko小胶质细胞的使用phrodo red标记的细菌生物颗粒和phrodo red淀粉样蛋白β的功能表征。将wt、1185 ht trem2 ko、1187 ho trem2 ko小胶质细胞在解冻后以15,000个活细胞/cm2的密度铺板在96孔板中的250μl mmm(图34a-b)或补充有仅mscf(图34c-d)或il-34(图34e-f)或il-34和mcsf的组合(图34g-h)的mdm基质(aka小胶质细胞基质培养基)中持续三天。用稀释的1μg/孔的调理或非调理phrodo生物颗粒(thermo fisher#a10010,每瓶2mg;储存在-20℃)(图34a、c、e、g)或phrodo淀粉样蛋白β(图34b、d、f、h)处理细胞。将板置于incucyte上,并在最多30小时的不同时间点拍摄吞噬作用的图像。wt以及工程化的小胶质细胞在解冻后显示出吞噬功能。动力学和吞噬作用效率在wt、1185 ht trem2 ko、1187 ho trem2 ko小胶质细胞之间不同。
80.图35:简化成熟培养基中小胶质细胞培养物的纯度。在补充有两种关键(il-34、mscf)细胞因子的组合的mmm或小胶质细胞基础培养基的存在下,第23天冷冻保存的野生型(wt)小胶质细胞在成熟培养基中的解冻后纯度。通过收获细胞在解冻后第3、7和14天定量纯度,并在分化过程结束时通过收获细胞确定cd45、cd33、trem2、cd11b、cx3cr1、p2ry12、tmem119、iba的纯度。通过流式细胞术对细胞进行细胞表面染色和细胞内标志物染色。冷冻保存的小胶质细胞在补充有mscf和il-34的成熟培养基中保持活力和纯度。这种简化的培养基对于冷冻保存的小胶质细胞与神经元和星形胶质细胞的用于开发大脑类器官trem的共培养应用会非常有价值。
81.图36a-36b:(图36a)描述使用冷冻保存的小胶质细胞的筛选实验的示意图。(图36b)筛选中测试的化合物的表。
82.图37a-37d:使用gw501516(图37a)、leucettine l41(图37b)、白皮杉醇(图37c)和azeliragon(图37d)的小胶质细胞筛选实验的结果。
83.图38a-38d:使用j147(图38a)、二丁酰-camp(图38b)、伊拉地平(图38c)和贝沙罗
汀(图38d)的小胶质细胞筛选实验的结果。
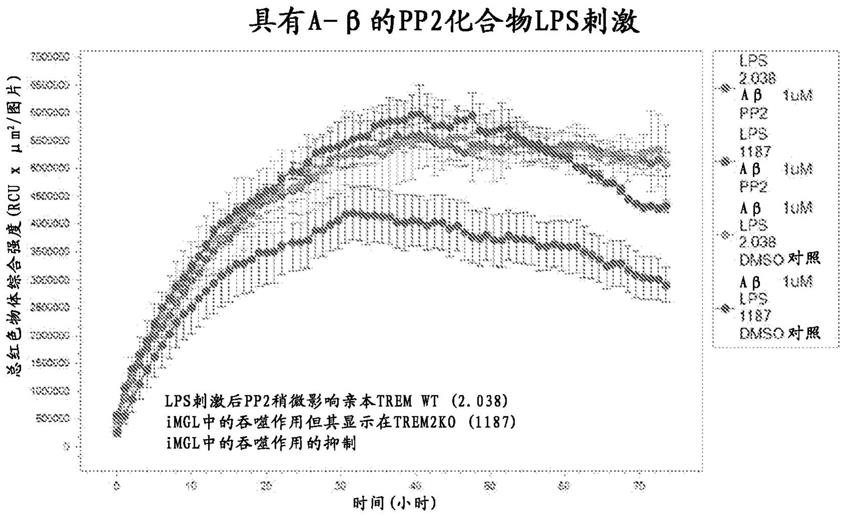
84.图39a-39e:使用sb-431542(图39a)、sp600125(图39b)、gw2580(图39c)、pp2(图39d)和sb239063(图39e)的小胶质细胞筛选实验的结果。
85.图40a-40d:在具有lps刺激的情况下使用gw501516(图40a)、leucettine l41(图40b)、白皮杉醇(图40c)和azeliragon(图40d)的小胶质细胞筛选实验的结果。
86.图41a-41e:在具有lps刺激的情况下使用j147(图41a)、二丁酰-camp(图41b)、伊拉地平(图41c)、贝沙罗汀(图41d)和sb-43152(图41e)的小胶质细胞筛选实验的结果。
87.图42a-42d:在具有lps刺激的情况下使用sp600125(图41a)、gw2580(图42b)、pp2(图42c)和sb239063(图42d)的小胶质细胞筛选实验的结果。
88.图43:小胶质细胞筛选实验结果的总结。
89.图44a-44g:(图44a)具有atp/bzatp所有迹线的小胶质细胞-比率。(图44b)具有atp/bzatp样品迹线的小胶质细胞-比率。(图44c)小胶质细胞对bzatp的反应。(图44d)小胶质细胞中对adp的差异反应。(图44e)在p2x7拮抗剂az11645373的存在下使用100μmbzatp的响应。(图44f)在p2x7拮抗剂a438079的存在下使用100μmbzatp的响应。(图44g)剂量依赖性反应以证明在azd1283(p2y
12
受体的有效拮抗剂)的存在下小胶质细胞中的功能性adp依赖性反应。
90.图45a-45b:使用simple step elisa(abcam)从在解冻后第3天(图45a)或第7天(图45b)收集的条件培养基定量源自anh和疾病相关小胶质细胞的冷冻保存的小胶质细胞中可溶性trem2的释放。将anh和dam相关的小胶质细胞解冻并在96孔primaria板中的成熟培养基中以相同的密度铺板。在解冻后第3天和解冻后第7天收集用过的培养基。在解冻后的第3天和第5天,培养物用新鲜成熟培养基进行半补料。
91.图46a-46j:来源于来自一组ipsc供体的anh和疾病相关小胶质细胞的冷冻保存的小胶质细胞释放的细胞因子和趋化因子。第23天冷冻保存的小胶质细胞在mdm培养基中解冻,并以50,000个细胞/孔铺板到primaria 96孔板上。在开始用100ng/ml lps或il-4 dbu-camp刺激之前,将细胞铺板三天。刺激一式三份进行,持续24小时。将上清液离心以去除细胞和碎片,并立即置于-20℃。在多重luminex测定上分析上清液。在用lps刺激后,m1分析物(图46a)、m2分析物(图46b)、白介素(图46c)、趋化因子(图46d)和其他分析物(图46e)的释放。在用il-4 dbu-camp刺激后,m1分析物(图46f)、m2分析物(图46g)、白介素(图46h)、趋化因子(图46i)和其他分析物(图46j)的释放。
92.图47a-47j:使用phrodo标记的金黄色葡萄球菌生物颗粒和淀粉样蛋白β证明ahn和疾病相关小胶质细胞的吞噬功能:将冷冻保存的ahn和疾病相关小胶质细胞以384孔聚-d-赖氨酸板的每孔5,000个细胞铺板。将金黄色葡萄球菌生物颗粒以0.5μg/ml的浓度添加到每个孔中,淀粉样蛋白β以1μm/孔的浓度添加。使用incucyte活细胞分析系统,使用总红色物体综合强度定量金黄色葡萄球菌生物颗粒和淀粉样蛋白β的吞噬动力学。(图47a)(anh,金黄色葡萄球菌),(图47b)(anh,β淀粉样蛋白),(图47c)(r47h相对anh,金黄色葡萄球菌),(图47d)(r47h相对anh,淀粉样蛋白β)、(图47e)(cd33相对anh、金黄色葡萄球菌)、(图47f)(cd33相对anh、淀粉样蛋白β)、(图47g)(abca7相对anh、金黄色葡萄球菌)、(图47h)(abca7相对anh,淀粉样蛋白β),(图47i)(apoe同种型相对anh,金黄色葡萄球菌),(图47j)(apoe同种型相对anh,淀粉样蛋白β)。
93.图:48a-48d:(图48a)在不使用双重smad抑制的情况下从ipsc产生神经前体细胞(npc)的方法的示意图。描述了所涉及的各个步骤以及所用培养基的组成。(图48b)三个ipsc系中npc的出现的动力学的总结。在分化过程的不同天数,多能性标志物的减少和npc特异性标志物的出现。通过细胞表面染色进行纯度的定量和通过流式细胞术进行细胞内染色(图48c)。来源于培养物中的多次传代的npc的星形胶质细胞的染色。通过流式细胞术通过细胞表面和细胞内染色定量星形胶质细胞的纯度。(图48d)将npc分化为神经元并通过细胞内流式细胞术定量末期神经元的纯度。
94.图49:与ipsc衍生的细胞谱系相容的表面的总结。
95.示例性实施方案的描述
96.在某些实施方案中,本公开内容提供用于从诱导多能干细胞(ipsc)产生多谱系细胞(例如内皮细胞、间充质干细胞(msc)和造血前体细胞(hpc))的方法。通常,该方法包括通过使用带电表面将ipsc分化为各种谱系细胞。具体而言,分化方法可以在不存在细胞外基质(ecm)蛋白的情况下进行,并允许通过传代进行自我净化,例如用于产生msc和内皮细胞。
97.在进一步的实施方案中,提供了用于将内皮细胞分化为脑微血管内皮细胞(bmec)或淋巴管内皮细胞、将msc分化为周细胞以及将hpc分化为小胶质细胞的方法。该过程允许有效生成内皮细胞和msc,而无需纯化例如macs纯化或使用ecm蛋白。该过程可以适应于良好生产规范(gmp)的合规性。
98.内皮细胞构成了人体中相互连接的细胞的网络,这些细胞沿血管、淋巴管排列并形成毛细血管。内皮细胞调节营养物质的流动并产生和响应不同的生物活性分子,并在用于针对血管毒性、血管通透性和治疗用途(包括治疗组织缺血和生物工程化移植物)筛选化合物和药物的工具中提供潜在用途。已经有许多方案用于衍生内皮细胞。几乎所有过程都需要使用cd31微珠进行磁激活细胞分选(mac)分离,以生成内皮细胞的纯培养物。
99.在某些实施方案中,本公开内容提供通过两步过程从ipsc(例如附加型重编程的ipsc)产生纯内皮细胞群的方法。ipsc细胞可以在带正电的胺表面上转化为造血祖细胞(hpc),然后在带负电荷的羧基表面的存在下进一步增殖和随后纯化内皮细胞。内皮细胞可以由造血内皮细胞衍生而无需任何macs纯化步骤。这些细胞以高纯度表达cd31/cd144/cd105,并能够繁殖以保持纯度并在较早期和晚期传代时冷冻保存。
100.从成人组织中分离的间充质干细胞(msc)能够在体外增殖并保持其多能性,使它们成为再生医学的有吸引力的细胞来源。然而,在目前的制备方案下,自我更新的可用性和能力是有限的。ipsc现在为msc提供了一种替代的、类似的细胞来源。因此,本公开内容的某些实施方案提供了通过使用gmp相容条件在带正电的胺表面上启动中胚层分化来从ipsc(例如附加型重编程的ipsc)分化msc的方法。通过该过程衍生的msc表达msc谱系的所有纯度标志物,并显示出自我更新和多能性。这些细胞可以按比例增加用于临床应用。
101.在进一步的实施方案中,本公开内容提供了用于将msc分化为周细胞(pc)的方法。周细胞(也称为壁细胞)包裹着血液微血管(即毛细血管、小动脉和小静脉),并通常被理解为在血管生成中具有组织或结构作用。多个标准(包括位置、形态、基因或蛋白质表达模式以及血管周围的密度)用于鉴定未成熟和成熟的周细胞。一般而言,可基于已知的周细胞分子标志物例如但不限于pdgfrβ、结蛋白(des)、cd13(anpep;丙氨酰(膜)氨肽酶)、α-sma、rgs5(g蛋白信号传导调节剂5)、ng2(也称为cspg4;硫酸软骨素蛋白多糖4)、cd248(内皮唾
液酸蛋白)、ang-1、cd146、cd44、cd90和cd13的表达来鉴定根据本文提供的方法获得的周细胞。
102.小胶质细胞是中枢神经系统的先天免疫细胞,在大脑发育、体内平衡和免疫调节中发挥关键作用。它们很难从人胎儿和初生组织中获得。因此,本公开内容的进一步实施方案提供了在确定的条件下从hpc(例如附加型重编程的icell hpc)产生、表征和冷冻保存人ipsc衍生的小胶质细胞(imgl)的方法。冷冻保存的imgl可保持纯度、分泌免疫调节细胞因子和吞噬phrodo red标记的细菌生物颗粒和淀粉样蛋白β聚集体。产生基本上无限数量的imgl的能力为加速对小胶质细胞在正常和疾病状态中的作用的人神经科学研究提供了巨大的希望。
103.本文进一步提供了在trem2、mecp2和/或scna中具有破坏的小胶质细胞。这些源自患者衍生的ipsc的小胶质细胞提供了一种体外工具来创建更准确的模型,以了解2d或3d类器官系统中人小胶质细胞、神经元、星形胶质细胞之间的复杂相互作用并模拟神经元疾病。
104.本文还提供了在不抑制smad信号传导的情况下将ipsc分化为神经前体细胞(npc)的方法。这些npc可以与ipsc衍生的小胶质细胞共培养,以产生长期共培养测定以在来源于正常和/或疾病特异性ipsc细胞的盘中模拟人脑发育和神经谱系、小胶质细胞、内皮细胞、周细胞和星形胶质细胞之间的复杂的细胞间相互作用。ipsc可以在分化开始之前维持在缺氧条件下以生成npc。为了启动神经前体分化,ipsc可以在存在rock抑制剂或blebbistatin的情况下铺板在ecm包被的表面上。细胞可以在接下来的48小时内在不存在rock抑制剂的情况下置于培养基中。接下来,细胞在补充有gsk3抑制剂的dmemf12培养基中预处理72小时,其中在常氧条件下每天更换培养基。细胞可以在预处理步骤结束时收获,然后以2d格式重新铺板回ecm包被的板上,或在rock抑制剂或blebbistatin的存在下使用超低附着板或旋转瓶作为3d聚集体重新铺板。在接下来的8天内,在常氧条件下,培养物可以每隔一天补料补充有n2的e6培养基以产生npc。在图45a中概述涉及生成npc的不同步骤。
105.通过本方法产生的细胞可用于疾病建模、药物发现和再生医学。本文还提供了使用本发明的细胞(例如,msc、内皮细胞、神经前体细胞和周细胞)来产生脑类器官或血脑屏障(bbb)模型的方法。
106.i.定义
107.如本文说明书所用的,“一个(a)”或“一种(an)”可表示一个或多个/一种或多种。如本文在权利要求中使用的,当与词“包含”结合使用时,词“一个”或“一种”可以表示一个或多于一个/一种或多于一种。
108.除非明确指出仅指替代方案或替代方案是相互排斥的(但是本公开内容支持仅指替代方案的定义和“和/或”),否则权利要求中使用的术语“或”用于表示“和/或”。如本文所用,“另一个”可表示至少第二个或更多个。
109.术语“基本上”应理解为方法或组合物仅包括指定的步骤或材料,以及不实质上影响那些方法和组合物的基本和新颖特征的那些步骤或材料。
110.如本文所用,“基本上不含”指定物质或材料的组合物或培养基含有≤30%、≤20%、≤15%,更优选≤10%,甚至更优选≤5%,或最优选≤1%的该物质或材料。
111.如本文所用,术语“大体上”或“大约”可用于修饰可允许变化而不导致与其相关的基本功能发生变化的任何定量比较、值、测量或其他表示。
112.术语“约”通常是指在使用测量所述值的标准分析技术确定的所述值的标准偏差内。这些术语也可以指加上或减去所述值的5%。
113.如本文所用,就指定组分而言,“基本上不含”在本文中用于表示没有任何指定组分被有意地配制到组合物中和/或仅作为污染物或以痕量存在。因此,由组合物的任何非故意污染导致的指定组分的总量远低于0.05%,优选低于0.01%。最优选的是这样的组合物,其中用标准分析方法无法检测到指定组分的量。
[0114]“无饲养层”或“不依赖饲养层”在本文中用于指补充有细胞因子和生长因子(例如,tgfβ、bfgf、lif)作为饲养细胞层的替代物的培养物。因此,“无饲养层”或不依赖饲养层的培养系统和培养基可用于培养和维持处于未分化和增殖状态的多能细胞。在一些情况下,无饲养层培养使用基于动物的基质(例如matrigel
tm
)或在基底例如纤连蛋白、胶原蛋白或玻连蛋白上生长。这些方法允许人干细胞保持基本上未分化状态,而无需小鼠成纤维细胞“饲养层”。
[0115]“饲养层”在本文中被定义为细胞的涂层,例如在培养皿的底部。饲养细胞能够将营养物质释放到培养基中,并提供其他细胞(如多能干细胞)能够附着的表面。
[0116]
术语“成分确定的”或“成分完全确定的”,当与培养基、细胞外基质或培养条件相关使用时,是指其中化学组分和几乎所有组分的量是已知的培养基、细胞外基质或培养条件。例如,成分确定的培养基不包含不确定的因子,例如胎牛血清、牛血清白蛋白或人血清白蛋白。通常,成分确定的培养基包括基础培养基(例如杜氏改良伊格尔培养基(dmem)、f12或roswell park memorial institute培养基(rpmi)1640,含有氨基酸、维生素、无机盐、缓冲液、抗氧化剂和能源),其补充有重组白蛋白、化学成分确定的脂质和重组胰岛素。成分完全确定的培养基的一个例子是essential 8
tm
培养基。
[0117]
对于与人细胞一起使用的培养基、细胞外基质或培养系统,术语“无异种(xf)”是指其中所使用的材料不具有非人动物来源的条件。
[0118]“治疗”或“医治”包括(1)抑制经历或表现出疾病的病理学或症状学的受试者或患者的疾病(例如,阻止病理学和/或症状学的进一步发展),(2)改善经历或表现出疾病的病理学或症状学的受试者或患者的疾病(例如,逆转病理学和/或症状学),和/或(3)实现经历或表现出疾病的病理学或症状学的受试者或患者的疾病的任何可测量的减少。
[0119]“预防性治疗”包括:(1)降低或减轻可能有患病风险和/或易患疾病但尚未经历或表现出疾病的任何或所有病理学或症状学的受试者或患者发展疾病的风险,和/或(2)减缓可能有患病风险和/或易患疾病但尚未经历或表现出疾病的任何或所有病理学或症状学的受试者或患者中疾病的病理学或症状学的发作。
[0120]
如本文所用,术语“患者”或“受试者”是指活的哺乳动物生物体,例如人、猴、牛、绵羊、山羊、狗、猫、小鼠、大鼠、豚鼠或其转基因物种。在某些实施方案中,患者或受试者是灵长类动物。人患者的非限制性实例是成人、青少年、婴儿和胎儿。
[0121]
在本说明书和/或权利要求中使用的术语“有效”是指足以实现期望的、预期的或意图的结果。当在用化合物治疗患者或受试者的上下文中使用时,“有效量”、“治疗有效量”或“药学上有效量”是指当施用至受试者或患者以治疗或预防疾病时足以影响疾病的这种治疗或预防的量。
[0122]
如本文中一般使用的,“药学上可接受的”是指在合理医学判断范围内适合用于与
人和动物的组织、器官和/或体液接触而没有过度的毒性、刺激、过敏反应或与合理的收益/风险比相应的其他问题或并发症的那些化合物、材料、组合物和/或剂型。
[0123]“诱导多能干细胞(ipsc)”是通过表达或诱导表达因子(在本文中称为重编程因子)的组合来重编程体细胞而产生的细胞。ipsc可以使用胎儿、产后、新生儿、青少年或成人体细胞生成。在某些实施方案中,可用于将体细胞重编程为多能干细胞的因子包括例如oct4(有时称为oct3/4)、sox2、c-myc、klf4、nanog和lin28。在一些实施方案中,通过表达至少两种重编程因子、至少三种重编程因子或四种重编程因子来重编程体细胞以将体细胞重编程为多能干细胞。
[0124]
术语“细胞外基质蛋白”是指为周围细胞提供结构和生化支持的分子。细胞外基质蛋白可以是重组的,并且还指其片段或肽。例子包括胶原蛋白和硫酸肝素。
[0125]“三维(3-d)培养”是指人工创造的环境,其中允许生物细胞在所有三个维度上生长或与其周围环境相互作用。3-d培养物能够在各种细胞培养容器(例如生物反应器、其中细胞能够生长成球体的小胶囊或非贴壁培养板)中生长。在特定方面,3-d培养是无支架的。相反,“二维(2-d)”培养是指贴壁表面上的细胞培养物例如单层。
[0126]
如本文所用,基因的“破坏”是指与不存在破坏的情况下的基因产物的表达水平相比,在细胞中由主题基因编码的一种或多种基因产物的表达的消除或减少。示例性基因产物包括基因编码的mrna和蛋白质产物。在一些情况下,破坏是暂时的或可逆的,而在其他情况下则是永久性的。在一些情况下,破坏是功能性或全长蛋白质或mrna的破坏,尽管事实上可能产生截短或无功能的产物。在本文的一些实施方案中,与表达相反,基因活性或功能被破坏。基因破坏通常由人工方法诱导,即通过添加或引入化合物、分子、复合物或组合物,和/或通过破坏基因的核酸或与基因相关的核酸,例如在dna水平上。基因破坏的示例性方法包括基因沉默、敲低、敲除和/或基因破坏技术,例如基因编辑。例子包括反义技术,例如rnai、sirna、shrna和/或核酶,它们通常导致表达的瞬时减少,以及导致靶向基因失活或破坏的基因编辑技术,例如通过诱导断裂和/或同源重组。例子包括插入、突变和缺失。破坏通常导致基因编码的正常或“野生型”产物的表达的抑制和/或完全缺失。此类基因破坏的实例是基因或基因的一部分的插入、移码和错义突变、缺失、敲入和敲除,包括整个基因的缺失。此类破坏可发生在编码区,例如,在一个或多个外显子中,导致无法产生全长产物、功能性产物或任何产物,例如通过插入终止密码子。此类破坏也可以通过启动子或增强子或其他影响转录激活的区域的破坏而发生,以阻止基因的转录。基因破坏包括基因靶向,包括通过同源重组的靶向基因失活。
[0127]
ii.ipsc分化方法
[0128]
a.hpc
[0129]
可以通过本领域已知的方法将ipsc分化为hpc,例如美国专利号8,372,642中描述的方法,该专利通过引用并入本文。在一种方法中,bmp4、vegf、flt3配体、il-3和gm-csf的组合可用于促进造血分化。在某些实施方案中,将细胞培养物依次暴露于第一培养基以制备用于分化的ipsc,包含bmp4、vegf和fgf的第二培养基,然后在包含flt3配体、scf、tpo、il-3和il-6的第三培养基中培养,可以将多能细胞分化为hpc和造血细胞。第二成分确定培养基还可以包含肝素。此外,在含有bmp4和vegf的培养基中加入fgf-2(50ng/ml)可以提高从多能细胞生成造血前体细胞的效率。此外,在第一成分确定培养基中加入糖原合酶激酶3
(gsk3)抑制剂(例如chir99021、bio和sb-216763)可以进一步提高hpc的产生。
[0130]
通常,多能细胞分化成造血前体细胞可以使用确定的或不确定的条件进行。应当理解,在意欲将所得细胞施用于人受试者的实施方案中,确定的条件通常是优选的。造血干细胞可以在确定的条件下(例如,使用tesr培养基)从多能干细胞衍生,并且造血细胞可以从多能细胞衍生的胚状体产生。在其他实施方案中,多能细胞可在op9细胞或小鼠胚胎成纤维细胞上共培养并随后分化。
[0131]
作为分化过程的一部分,可以允许多能细胞形成胚状体或聚集体。形成“胚状体”(eb)或生长细胞的簇以诱导分化通常涉及人多能干细胞在体外聚集成eb并允许人多能干细胞自发和随机分化为代表内胚层、外胚层和中胚层起源的多个组织类型。因此,三维eb可用于产生一定比例的造血细胞和内皮细胞。
[0132]
为了促进聚集体形成,可以将细胞转移到低附着板用于在无血清分化(sfd)培养基中孵育过夜,该培养基由75%imdm(gibco)、不含ra补充剂的补充有0.05%n2和1%b-27的25%ham's modified f12(cellgro)、200mm 1-谷氨酰胺、0.05mg/ml抗坏血酸-2-磷酸镁盐(asc 2-p)(wako)和4.5x10-4
mtg组成。第二天可以从每个孔中收集细胞并离心。然后对于分化的前四天可以将细胞重新悬浮在“eb分化培养基”中,该培养基由补充有约50ng/ml骨形态发生因子(bmp4)、约50ng/ml血管内皮生长因子(vegf)和50ng/ml zb fgf的sfd基础培养基组成。细胞每48小时进行半补料。在分化的第五天,培养基更换为由补充有50ng/ml干细胞因子(scf)、约50ng/ml flt-3配体(flt-3l)、50ng/ml白介素-6(il-6)、50ng/ml白介素-3(il-3)、50ng/ml血小板生成素(tpo)的sfd培养基组成的第二培养基。细胞每48小时用新鲜分化培养基进行半补料。培养基更换是通过将分化培养物以300g离心5分钟并从分化培养物中吸出一半体积并用新鲜培养基补充它来进行的。在某些实施方案中,eb分化培养基可包括约bmp4(例如约50ng/ml)、vegf(例如约50ng/ml)和任选地fgf-2(例如约25-75ng/ml或约50ng/ml)。可以吸出上清液并更换为新鲜的分化培养基。或者,细胞可以每两天用新鲜培养基进行半补料。细胞可以在分化过程中的不同时间点收获。
[0133]
可以使用成分确定的培养基从多能干细胞培养hpc。使用成分确定的培养基将多能细胞分化为造血cd34
干细胞的方法描述于例如美国申请12/715,136中,其全部内容通过引用并入本文。预计这些方法可用于本公开内容。
[0134]
例如,可以使用成分确定的培养基来诱导造血cd34
分化。成分确定的培养基可包含生长因子bmp4、vegf、flt3配体、il-3和/或gmcsf。多能细胞可以在包含bmp4、vegf和任选地fgf-2的第一成分确定培养基中培养,然后在包含(flt3配体、il-3和gmcsf)或(flt3配体、il-3、il-6和tpo)的第二培养基中培养。第一和第二培养基还可包含scf、il-6、g-csf、epo、fgf-2和/或tpo中的一种或多种。基本上缺氧条件(例如,低于20%o2)可能进一步促进造血或内皮分化。
[0135]
细胞可以通过机械或酶促工具(例如,使用胰蛋白酶或tryple
tm
)基本上个体化。rock抑制剂(例如h1152或y-27632)也可以包含在培养基中。预计这些方法可以使用例如机器人自动化来自动化。
[0136]
在某些实施方案中,可使用基本上缺氧条件来促进多能细胞分化成造血祖细胞。本领域技术人员将认识到,小于约20.8%的大气氧含量将被认为是缺氧的。培养中的人细胞能够在与环境空气相比氧含量降低的大气条件下生长。这种相对缺氧可以通过减少暴露
于培养基的大气氧来实现。胚胎细胞通常在降低氧条件(通常在大约1%和大约6%的大气氧之间)下在体内发育,其中二氧化碳处于环境水平。不希望受理论束缚,预计缺氧条件可以模拟某些胚胎发育条件的一个方面。如以下实施例所示,在某些实施方案中可以使用缺氧条件来促进诱导的多能细胞进一步分化为分化程度更高的细胞类型,例如hpc。
[0137]
以下缺氧条件可用于促进多能细胞分化为造血祖细胞。在某些实施方案中,小于约20%、小于约19%、小于约18%、小于约17%、小于约16%、小于约15%、小于约14%、小于约13%、小于约12%、小于约11%、小于约10%、小于约9%、小于约8%、小于约7%、小于约6%、小于约5%、约5%、约4%、约3%、约2%或约1%的大气氧含量可用于促进分化为造血前体细胞。在某些实施方案中,缺氧大气包含约5%的氧气。
[0138]
不管在任何给定的造血祖细胞扩增中使用的具体培养基如何,所用培养基优选补充有浓度为约0.1ng/ml至约500ng/ml,更通常为10ng/ml至100ng/ml的至少一种细胞因子。合适的细胞因子包括但不限于c-kit配体(kl)(也称为青灰因子(sti)、肥大细胞生长因子(mgf)和干细胞因子(scf))、il-6、g-csf、il-3、gm-csf、il-1α、il-11mip-1α、lif、c-mpl配体/tpo和flk2/flk3配体(flt2l或flt3l)。特别地,培养将包含scf、flt3l和tpo中的至少一种。更具体地说,培养将包含scf、flt3l和tpo。
[0139]
在一个实施方案中,细胞因子包含在培养基中并通过培养基灌注补充。或者,当使用生物反应器系统时,细胞因子可以作为浓缩溶液通过单独的入口单独添加,无需培养基灌注。当细胞因子在没有灌注的情况下加入时,它们通常以等于生物反应器中体积的十分之一到1/100的量作为10x到100x溶液添加,并且大约每2到4天添加新鲜的细胞因子。此外,除了灌注培养基中的细胞因子外,还可以单独添加新鲜浓缩的细胞因子。
[0140]
示例性hpc分化方法
[0141]
2d hpc分化:ipsc可在e8的存在下维持在matrigel
tm
或玻连蛋白上并适应缺氧至少5-10代。将细胞从亚汇合ipsc中分开,并以25万个细胞/孔的密度在补充有5um blebbistatin的无血清的成分确定的(sfd)培养基的存在下铺板到胺培养皿上。铺板后24小时,将补充有50ng/ml bmp4、vegf和fgf2的sfd培养基添加到培养物中。第二天,更换新鲜培养基以去除blebbistatin。在分化过程的第五天,将细胞置于含有50ng/ml flt-3配体、scf、tp0、il3和il6以及5u/ml肝素的培养基中。在整个分化过程中,细胞每48小时进行补料。整个过程在缺氧条件下和在带电的胺板上进行。hpc通过cd43/cd34细胞的存在和cfu进行定量。
[0142]
3d hpc分化:细胞从亚汇合的ipsc分开,并在补充有5μm blebbistatin或1μm h1152的无血清(sfd)培养基的情况下以每毫升25-50万个细胞的密度铺板到旋转瓶中。铺板后24小时更换补充有50ng/ml bmp4、vegf和fgf2的sfd培养基。在分化过程的第五天,将细胞置于含有50ng/ml flt-3配体、scf、tp0、il3和il6以及5-10u/ml肝素的培养基中。在整个分化过程中,细胞每48小时进行补料。整个过程在缺氧条件下进行。通过cd43/cd34的存在定量hpc。hpc是使用cd34珠进行macs分选的。
[0143]
b.基因破坏
[0144]
在某些方面,在细胞例如psc(例如esc或ipsc)中trem2、mecp2和/或scna基因表达、活性或功能被破坏。在一些实施方案中,基因破坏是通过在基因中实现破坏(例如敲除、插入、错义或移码突变,例如双等位基因移码突变、基因的全部或部分(例如一个或更多的
外显子或其部分)的缺失和/或敲入)来进行的。例如,可以通过序列特异性或靶向核酸酶(包括dna结合靶向核酸酶,例如锌指核酸酶(zfn)和转录激活因子样效应核酸酶(talen),以及rna引导的核酸酶,例如crispr-相关核酸酶(cas),专门设计用于靶向基因的序列或其部分)进行破坏。
[0145]
在一些实施方案中,基因的表达、活性和/或功能的破坏是通过破坏基因来进行的。在一些方面,基因被破坏以使其表达与在不存在基因破坏或不存在引入以实现破坏的成分的情况下的表达相比降低至少或约20、30或40%,通常至少或约50、60、70、80、90或95%。
[0146]
在一些实施方案中,破坏是瞬时的或可逆的,使得基因的表达在以后恢复。在其他实施方案中,破坏不是可逆的或暂时的,例如是永久性的。
[0147]
在一些实施方案中,通常以靶向方式通过诱导基因中的一个或多个双链断裂和/或一个或多个单链断裂来进行基因破坏。在一些实施方案中,双链或单链断裂由核酸酶例如内切核酸酶例如基因靶向核酸酶产生。在一些方面,在基因的编码区例如在外显子中诱导断裂。例如,在一些实施方案中,诱导发生在编码区的n-末端部分附近,例如在第一个外显子中、在第二个外显子中或在随后的外显子中。
[0148]
在一些方面,双链或单链断裂通过细胞修复过程(例如通过非同源末端连接(nhej)或同源定向修复(hdr))进行修复。在一些方面,修复过程容易出错并且导致基因的破坏,例如移码突变,例如双等位基因移码突变,这可以导致基因的完全敲除。例如,在一些方面,破坏包括诱导缺失、突变和/或插入。在一些实施方案中,破坏导致早期终止密码子的存在。在一些方面,插入、缺失、易位、移码突变和/或提前终止密码子的存在导致基因的表达、活性和/或功能的破坏。
[0149]
在一些实施方案中,使用反义技术实现基因破坏,例如通过rna干扰(rnai)、短干扰rna(sirna)、短发夹(shrna),和/或使用核酶用于选择性地抑制或阻遏基因的表达。sirna技术是rnai,其利用具有与从基因转录的mrna的核苷酸序列同源的序列和与核苷酸序列互补的序列的双链rna分子。sirna通常与从基因转录的mrna的一个区域同源/互补,或者可以是包括多个与不同区域同源/互补的rna分子的sirna。在一些方面,sirna包含在多顺反子构建体中。在特定方面,sirna抑制来自内源性mrna的野生型和突变型蛋白质翻译。
[0150]
在一些实施方案中,使用dna靶向分子实现破坏,例如dna结合蛋白或dna结合核酸,或包含它们的复合物、化合物或组合物,其特异性结合或杂交基因。在一些实施方案中,dna靶向分子包含dna结合结构域,例如锌指蛋白(zfp)dna结合结构域、转录激活子样蛋白(tal)或tal效应子(tale)dna结合结构域,成簇的规则间隔短回文重复序列(crispr)dna结合结构域,或来自兆核酸酶的dna结合结构域。锌指、tale和crispr系统结合结构域能够被工程化以结合预定的核苷酸序列,例如通过工程化(改变一个或多个氨基酸)天然存在的锌指或tale蛋白的识别螺旋区域。工程化dna结合蛋白(锌指或tale)是非天然存在的蛋白质。设计的合理标准包括应用替代规则和计算机算法用于处理存储现有zfp和/或tale设计的信息和结合数据的数据库中的信息。参见,例如,美国专利号6,140,081;6,453,242;和6,534,261;也参见wo98/53058;wo98/53059;wo98/53060;wo02/016536和wo03/016496以及美国公开号2011/0301073。
[0151]
在一些实施方案中,dna靶向分子、复合物或组合包含dna结合分子和一个或多个
附加结构域,例如促进基因阻遏或破坏的效应结构域。例如,在一些实施方案中,基因破坏通过包含dna结合蛋白和异源调节结构域或其功能片段的融合蛋白进行。在一些方面,域包括例如转录因子域,例如激活子、阻遏物、共激活物、共阻遏物、沉默子、癌基因、dna修复酶及其相关因子和修饰剂、dna重排酶及其相关因子和修饰剂,染色质相关蛋白及其修饰物,例如激酶、乙酰化酶和去乙酰化酶,以及dna修饰酶,例如甲基转移酶、拓扑异构酶、解旋酶、连接酶、激酶、磷酸酶、聚合酶、内切核酸酶及其相关因子和修饰剂。参见,例如,美国专利申请公开号2005/0064474;2006/0188987和2007/0218528,其通过引用整体并入本文,提供关于dna结合结构域和核酸酶切割结构域的融合体的细节。在一些方面,附加结构域是核酸酶域结构。因此,在一些实施方案中,通过基因或基因组编辑促进基因破坏,使用工程化蛋白,例如核酸酶和含核酸酶的复合物或融合蛋白,包含与非特异性dna切割分子例如核酸酶融合或复合的序列特异性dna结合结构域。
[0152]
在一些方面,这些靶向嵌合核酸酶或含核酸酶的复合物通过诱导靶向双链断裂或单链断裂、刺激细胞dna修复机制包括容易出错的非同源末端连接(nhej)和同源定向修复(hdr)来进行精确的遗传修饰。在一些实施方案中,核酸酶是内切核酸酶,例如锌指核酸酶(zfn)、tale核酸酶(talen)和rna引导的内切核酸酶(rgen),例如crispr相关(cas)蛋白或兆核酸酶。
[0153]
在一些实施方案中,提供供体核酸,例如供体质粒或编码基因工程化抗原受体的核酸,并将其在引入dsb后通过hdr插入基因编辑位点。因此,在一些实施方案中,基因的破坏和抗原受体例如car的引入同时进行,由此基因部分地通过敲入或插入编码car的核酸而被破坏。
[0154]
在一些实施方案中,不提供供体核酸。在一些方面,引入dsb后nhej介导的修复导致插入或缺失突变,其可导致基因破坏,例如通过产生错义突变或移码。
[0155]
1.zfp和zfn
[0156]
在一些实施方案中,dna靶向分子包括与效应蛋白例如内切核酸酶融合的dna结合蛋白例如一种或多种锌指蛋白(zfp)或转录激活物样蛋白(tal)。实例包括zfn、tale和talen。
[0157]
在一些实施方案中,靶向dna的分子包含一种或多种以序列特异性方式与dna结合的锌指蛋白(zfp)或其结构域。zfp或其结构域是较大蛋白质内的蛋白质或结构域,其通过一个或多个锌指(结合结构域内的其结构通过锌离子的配位稳定的氨基酸序列区域)以序列特异性方式结合dna。术语锌指dna结合蛋白通常缩写为锌指蛋白或zfp。在zfp中有靶向特定dna序列的人工zfp结构域,通常长度为9-18个核苷酸,由个体指的组装产生。
[0158]
zfp包括其中单个指结构域的长度约为30个氨基酸并含有α螺旋的那些,该α螺旋含有通过锌与单个β转角的两个半胱氨酸配位的两个不变组氨酸残基,并且具有两个、三个、四个、五个或六个指。通常,zfp的序列特异性可以通过在锌指识别螺旋上的四个螺旋位置(-1、2、3和6)进行氨基酸取代来改变。因此,在一些实施方案中,zfp或含zfp的分子是非天然存在的,例如,被工程化为与选择的靶位点结合。
[0159]
在一些方面,mecp2的破坏是通过使基因中的第一靶位点与第一zfp接触从而破坏基因来进行的。在一些实施方案中,基因中的靶位点与包含六个指和调节结构域的融合zfp接触,从而抑制基因的表达。
[0160]
在一些实施方案中,接触步骤还包括使基因中的第二靶位点与第二zfp接触。在一些方面,第一和第二靶位点是相邻的。在一些实施方案中,第一和第二zfp共价连接。在一些方面,第一zfp是包含一个调节结构域或至少两个调节结构域的融合蛋白。
[0161]
在一些实施方案中,第一和第二zfp是融合蛋白,各自包含一个调节结构域或各自包含至少两个调节结构域。在一些实施方案中,调节结构域是转录阻遏物、转录激活物、内切核酸酶、甲基转移酶、组蛋白乙酰转移酶或组蛋白脱乙酰酶。
[0162]
在一些实施方案中,zfp由可操作地连接至启动子的zfp核酸编码。在一些方面,该方法进一步包括首先将核酸在脂质:核酸复合物中或作为裸核酸施用至细胞的步骤。在一些实施方案中,zfp由包含可操作地连接至启动子的zfp核酸的表达载体编码。在一些实施方案中,zfp由与诱导型启动子可操作连接的核酸编码。在一些方面,zfp由与弱启动子可操作连接的核酸编码。
[0163]
在一些实施方案中,靶位点在基因的转录起始位点的上游。在一些方面,靶位点与基因的转录起始位点相邻。在一些方面,靶位点与基因转录起始位点下游的rna聚合酶暂停位点相邻。
[0164]
在一些实施方案中,靶向dna的分子是或包含与dna切割结构域融合以形成锌指核酸酶(zfn)的锌指dna结合结构域。在一些实施方案中,融合蛋白包含来自至少一种lis型限制酶的切割结构域(或切割半结构域)和一个或多个锌指结合结构域,其可以被或可以不被工程化。在一些实施方案中,切割结构域来自lis型限制性内切核酸酶foki。foki通常催化dna的在一条链上距其识别位点9个核苷酸处和在另一条链上距其识别位点13个核苷酸处的双链切割。
[0165]
在一些实施方案中,zfn靶向工程化细胞中存在的基因。在一些方面,zfn有效地产生双链断裂(dsb),例如在基因编码区中的预定位点处。靶向的典型区域包括外显子、编码n末端区域、第一外显子、第二外显子的区域和启动子或增强子区域。在一些实施方案中,zfn的瞬时表达促进工程化细胞中靶基因的高效和永久破坏。特别地,在一些实施方案中,zfn的递送导致基因的永久性破坏,效率超过50%。
[0166]
许多基因特异性工程化锌指可商购获得。例如,sangamo biosciences(richmond,ca,usa)与sigma-aldrich(st.louis,mo,usa)合作开发了用于锌指构建的平台(compozr),使研究人员能够完全绕过锌指构建和验证并为数千种蛋白质提供特异性靶向锌指(gaj等人,trends in biotechnology,2013,31(7),397-405)。在一些实施方案中,使用或定制设计可商购获得的锌指。
[0167]
2.tal、tale和talen
[0168]
在一些实施方案中,dna靶向分子包含天然存在的或工程化的(非天然存在的)转录激活因子样蛋白(tal)dna结合结构域,例如在转录激活因子样蛋白效应物(tale)蛋白中,参见,例如,美国专利公开号2011/0301073,其全部内容通过引用并入本文。
[0169]
tale dna结合结构域或tale是包含一个或多个tale重复结构域/单元的多肽。重复结构域参与tale与其同源靶dna序列的结合。单个“重复单元”(也称为“重复序列”)的长度通常为33-35个氨基酸,并且与天然存在的tale蛋白内的其他tale重复序列显示出至少一些序列同源性。每个tale重复单元包含1或2个dna结合残基,构成重复可变双残基(rvd),通常位于重复序列的12和/或13位。已确定这些tale的dna识别的自然(典型)代码,使得第
12和13位的hd序列导致与胞嘧啶(c)的结合,ng与t结合,ni与a结合,nn与g或a结合和no与t结合,并且非经典(非典型)rvd也是已知的。参见美国专利公开号2011/0301073。在一些实施方案中,通过设计对靶dna序列具有特异性的tal阵列,tale可以靶向任何基因。靶序列通常以胸苷开始。
[0170]
在一些实施方案中,分子是dna结合内切核酸酶,例如tale核酸酶(talen)。在一些方面,talen是包含源自tale的dna结合结构域和切割核酸靶序列的核酸酶催化结构域的融合蛋白。
[0171]
在一些实施方案中,talen识别并切割基因中的靶序列。在一些方面,dna的切割导致双链断裂。在一些方面,断裂刺激同源重组或非同源末端连接(nhej)的速率。通常,nhej是一个不完善的修复过程,其通常导致在切割位点处的dna序列的变化。在一些方面,修复机制涉及通过直接重新连接(critchlow和jackson,1998)或通过所谓的微同源介导的末端连接重新连接两个dna末端的剩余部分。在一些实施方案中,经由nhej的修复导致小的插入或缺失并且可用于破坏并由此阻遏基因。在一些实施方案中,修饰可以是至少一个核苷酸的取代、缺失或添加。在一些方面,可以通过本领域众所周知的方法鉴定和/或选择其中发生切割诱导的诱变事件即与nhej事件连续的诱变事件的细胞。
[0172]
在一些实施方案中,组装tale重复序列以特异性靶向基因。已经构建了靶向18,740个人蛋白质编码基因的talen文库。定制设计的tale阵列可通过cellectis bioresearch(paris,france)、transposagen biopharmaceuticals(lexington,ky,usa)和life technologies(grand island,ny,usa)商购获得。
[0173]
在一些实施方案中,talen作为由一种或多种质粒载体编码的转基因被引入。在一些方面,质粒载体可包含选择标志物,其提供接受所述载体的细胞的鉴定和/或选择。
[0174]
3.rgen(crispr/cas系统)
[0175]
在一些实施方案中,使用一种或多种dna结合核酸进行破坏,例如通过rna引导的内切核酸酶(rgen)进行破坏。例如,可以使用成簇的规则间隔短回文重复序列(crispr)和crispr相关(cas)蛋白进行破坏。一般而言,“crispr系统”统称参与crispr相关(“cas”)基因的表达或指导其活性的转录物和其他元件,包括编码cas基因的序列、tracr(反式激活crispr)序列(例如,tracrrna或活性部分tracrrna)、tracr配对序列(包括在内源性crispr系统的上下文中的“同向重复序列”和tracrrna加工的部分同向重复序列)、引导序列(在内源性crispr系统的上下文中也称为“间隔区”)和/或来自crispr基因座的其他序列和转录物。
[0176]
crispr/cas核酸酶或crispr/cas核酸酶系统可以包括非编码rna分子(引导)rna(其序列特异性地结合dna)和具有核酸酶功能(例如,两个核酸酶结构域)的cas蛋白(例如,cas9)。crispr系统的一个或多个元件可源自i型、ii型或iii型crispr系统,例如源自包含内源性crispr系统的特定生物体,例如化脓性链球菌。
[0177]
在一些方面,将cas核酸酶和grna(包括对靶序列特异的crrna和固定的tracrrna的融合体)引入细胞。通常,grna的5'端的靶位点使用互补碱基配对将cas核酸酶靶向至靶位点例如基因。靶位点可基于其紧邻前间区序列邻近基序(pam)序列(例如通常为ngg或nag)的5'的位置来选择。在这方面,grna通过修饰引导rna的前20、19、18、17、16、15、14、14、12、11或10个核苷酸以对应于靶dna序列来靶向至所需序列。一般而言,crispr系统的特征
在于促进在靶序列位点形成crispr复合物的元件。通常,“靶序列”通常是指引导序列被设计成与之具有互补性的序列,其中靶序列和引导序列之间的杂交促进了crispr复合物的形成。不一定需要完全互补,只要有足够的互补性来引起杂交并促进crispr复合物的形成。
[0178]
crispr系统可以在靶位点诱导双链断裂(dsb),然后是如本文所讨论的破坏。在其他实施方案中,被视为“切口酶”的cas9变体用于在靶位点切割单链。可以使用成对的切口酶,例如,以提高特异性,每个切口酶由一对不同的grna靶向序列引导,使得在引入切口时,同时引入5'突出端。在其他实施方案中,催化失活的cas9与异源效应结构域例如转录阻遏物或激活物融合,以影响基因表达。
[0179]
靶序列可以包含任何多核苷酸,例如dna或rna多核苷酸。靶序列可以位于细胞的细胞核或细胞质中,例如在细胞的细胞器内。通常,可用于重组到包含靶序列的靶基因座中的序列或模板被称为“编辑模板”或“编辑多核苷酸”或“编辑序列”。在一些方面,外源模板多核苷酸可称为编辑模板。在一些方面,重组是同源重组。
[0180]
通常,在内源性crispr系统的上下文中,crispr复合物(包含与靶序列杂交并与一种或多种cas蛋白复合的引导序列)的形成导致在靶序列中或附近(例如在距靶序列的1、2、3、4、5、6、7、8、9、10、20、50或更多碱基对内)的一条或两条链的切割。tracr序列(其可包含野生型tracr序列的全部或部分或由其组成(例如野生型tracr序列的约或多于约20、26、32、45、48、54、63、67、85个或更多个核苷酸)也可以形成crispr复合物的一部分,例如通过沿着至少部分tracr序列与可操作地连接到引导序列的tracr配对序列的全部或部分杂交。tracr序列与tracr配对序列具有足够的互补性以杂交并参与crispr复合物的形成,例如在最佳比对时沿着tracr配对序列的长度至少50%、60%、70%、80%、90%、95%或99%的序列互补性。
[0181]
驱动crispr系统的一种或多种元件的表达的一种或多种载体可以被引入到细胞中,使得crispr系统的元件的表达指导crispr复合物在一个或多个靶位点处的形成。组分也可以作为蛋白质和/或rna递送至细胞。例如,cas酶、与tracr配对序列连接的引导序列和tracr序列可以各自与单独载体上的单独调节元件可操作地连接。或者,从相同或不同调节元件表达的两个或更多个元件可以组合在单个载体中,其中一个或多个额外载体提供未包括在第一载体中的crispr系统的任何组分。载体可包含一个或多个插入位点,例如限制性内切核酸酶识别序列(也称为“克隆位点”)。在一些实施方案中,一个或多个插入位点位于一个或多个载体的一个或多个序列元件的上游和/或下游。当使用多个不同的引导序列时,可以使用单个表达构建体将crispr活性靶向至细胞内的多个不同的对应靶序列。
[0182]
载体可以包含与编码crispr酶(例如cas蛋白)的酶编码序列可操作地连接的调节元件。cas蛋白的非限制性实例包括cas1、cas1b、cas2、cas3、cas4、cas5、cas6、cas7、cas8、cas9(也称为csn1和csx12)、cas10、csy1、csy2、csy3、cse1、cse2、csc1、csc2、csa5、csn2、csm2、csm3、csm4、csm5、csm6、cmr1、cmr3、cmr4、cmr5、cmr6、csb1、csb2、csb3、csx17、csx14、csx10、csx16、csax、csx3、csx1、csx15、csfl、csf2、csf3、csf4、它们的同系物或其修饰形式。这些酶是已知的;例如,化脓性链球菌cas9蛋白的氨基酸序列可以在swissprot数据库中找到,登录号为q99zw2。
[0183]
crispr酶可以是cas9(例如,来自化脓性链球菌或肺炎链球菌)。crispr酶可以在靶序列的位置例如在靶序列内和/或在靶序列的互补序列内指导一条或两条链的切割。载
体可以编码相对于相应的野生型酶突变的crispr酶,使得突变的crispr酶缺乏切割含有靶序列的靶多核苷酸的一条或两条链的能力。例如,来自化脓性链球菌的cas9的ruvci催化结构域中的天冬氨酸到丙氨酸的取代(d10a)将cas9从切割两条链的核酸酶转化为切口酶(切割单链)。在一些实施方案中,cas9切口酶可以与引导序列(例如,两个引导序列,其分别靶向dna靶标的有义链和反义链)组合使用。这种组合允许两条链都被切割并用于诱导nhej或hdr。
[0184]
在一些实施方案中,编码crispr酶的酶编码序列是密码子优化的以用于在特定细胞例如真核细胞中表达。真核细胞可以是特定生物体(例如哺乳动物,包括但不限于人、小鼠、大鼠、兔、狗或非人灵长类动物)或源自其的真核细胞。一般而言,密码子优化是指通过用在该宿主基因中更频繁或最频繁使用的密码子替换天然序列的至少一个密码子来修饰核酸序列以增强在感兴趣的宿主细胞中的表达同时保持天然氨基酸序列的过程。各种物种对特定氨基酸的某些密码子表现出特定的偏向性。密码子偏向性(生物体之间密码子使用的差异)通常与信使rna(mrna)的翻译效率相关,而信使rna(mrna)的翻译效率又被认为除了其他以外取决于翻译的密码子的特性和特定转移rna(trna)分子的可用性。细胞中所选trna的优势通常反映了肽合成中最常用的密码子。因此,可以基于密码子优化来定制基因以在给定生物体中进行最佳基因表达。
[0185]
一般而言,引导序列是与靶多核苷酸序列具有足够互补性以与靶序列杂交并指导crispr复合物与靶序列的序列特异性结合的任何多核苷酸序列。在一些实施方案中,当使用合适的比对算法最佳比对时,引导序列与其对应的靶序列之间的互补程度为约或大于约50%、60%、75%、80%、85%、90%、95%、97.5%、99%或更多。
[0186]
最佳比对可以使用用于比对序列的任何合适算法来确定,其非限制性实例包括smith-waterman算法、needleman-wunsch算法、基于burrows-wheeler变换的算法(例如burrows wheeler aligner)、clustal w、clustal x、blat、novoalign(novocraft technologies、eland(illumina,san diego,calif.)、soap(可在soap.genomics.org.cn获得)和maq(可在maq.sourceforge.net获得)。
[0187]
crispr酶可以是包含一个或多个异源蛋白质结构域的融合蛋白的一部分。crispr酶融合蛋白可以包含任何额外的蛋白质序列,并且任选地包含任何两个结构域之间的接头序列。可与crispr酶融合的蛋白质结构域的实例包括但不限于表位标签、报告基因序列和具有以下一种或多种活性的蛋白质结构域:甲基化酶活性、去甲基化酶活性、转录激活活性、转录抑制活性、转录释放因子活性、组蛋白修饰活性、rna切割活性和核酸结合活性。表位标签的非限制性实例包括组氨酸(his)标签、v5标签、flag标签、流感血凝素(ha)标签、myc标签、vsv-g标签和硫氧还蛋白(trx)标签。报告基因的实例包括但不限于谷胱甘肽-5-转移酶(gst)、辣根过氧化物酶(hrp)、氯霉素乙酰转移酶(cat)、β-半乳糖苷酶、β-葡萄糖醛酸酶、萤光素酶、绿色荧光蛋白(gfp)、hcred、dsred、青色荧光蛋白(cfp)、黄色荧光蛋白(yfp)和自发荧光蛋白,包括蓝色荧光蛋白(bfp)。crispr酶可以与编码结合dna分子或结合其他细胞分子的蛋白质或蛋白质片段的基因序列融合,所述其他细胞分子包括但不限于麦芽糖结合蛋白(mbp)、s-标签、lex a dna结合结构域(dbd)融合体、gal4a dna结合结构域融合体和单纯疱疹病毒(hsv)bp16蛋白融合体。
[0188]
c.带电细胞表面
[0189]
在一些实施方案中,本公开内容涉及用于细胞培养的带电表面。带电表面可以带正电,例如胺表面或含氮官能团,或带负电,例如羧基表面或含氧官能团。可以处理细胞表面以改变培养容器的表面电荷。
[0190]
在一些方面,表面是中性带电的,例如包含带负电荷和带正电的官能团的表面。例如,corning表面在聚苯乙烯表面上具有含氧(带负电)和含氮(带正电)官能团的独特混合物。当在传统tc表面上培养时,表面支持细胞的生长,这些细胞可能表现出较差的附着或有限的分化潜能。在一些方面,表面包括ula表面涂层。例如,康宁超低附着表面是一种共价结合的水凝胶层,其亲水且中性带电。由于蛋白质和其他生物分子通过疏水或离子相互作用被动吸附到聚苯乙烯表面,这种水凝胶通过这些力天然地抑制非特异性固定,从而抑制随后的细胞附着。该表面非常稳定、无细胞毒性、生物惰性且不可降解。其他可以支持从hpc生成小胶质细胞的例子包括:corning cellbind培养(美国专利6,617,152)使用更高能量的微波等离子体将更多的氧掺入到聚苯乙烯表面上,使其与传统的等离子体或电晕放电处理的表面相比更具亲水性(可湿性),同时增加了表面的稳定性。corning synthemax自包被底物是一种含有rgd基序和侧翼序列的独特的、不含动物成分的、基于玻连蛋白的合成肽。该合成肽与聚合物主链共价结合用于被动包被、定向和呈递肽以用于最佳的细胞结合和信号传导。
[0191]
细胞培养表面可以包被有等离子体聚合膜。等离子体聚合的来源是一种或多种单体。有用的可聚合单体可包括不饱和有机化合物,例如烯胺、卤代烯烃、烯羧酸和羧酸盐、烯腈化合物、氧化烯烃和烯烃。在一些实施方案中,烯烃可包括乙烯基和烯丙基形式。在其他实施方案中,可以使用环状化合物,例如环己烷、环戊烷和环丙烷。
[0192]
本领域技术人员将认识到,可以利用各种等离子体聚合技术将一种或多种单体沉积到细胞培养表面上。优选地,将带正电的聚合膜沉积在表面上。如本领域技术人员将理解的,取决于与其一起使用的蛋白质,等离子体聚合表面可具有负电荷。胺优选用作聚合物的单体来源。在一些实施方案中,等离子体聚合单体使用等离子体源制造以产生气体放电,其提供能量以引发气态单体的聚合并且允许薄聚合物膜沉积在培养容器上。可以使用环状化合物,其可以包括通过辉光放电方法产生的气体等离子体。这些环状化合物的衍生物例如1,2-二氨基环己烷通常也可在气体等离子体中聚合。
[0193]
可以使用可聚合单体的混合物。此外,可聚合单体可与通常认为其本身不可聚合的其他气体(例如氩气、氮气和氢气)混合。
[0194]
考虑了可使用可用于贴壁培养的任何培养容器。本公开内容考虑的优选细胞培养容器配置包括多孔板(例如6孔板、12孔板和24孔板)、培养皿(例如皮氏培养皿)、试管、培养瓶、滚瓶、管或摇瓶等。
[0195]
用于细胞培养表面的材料可以包括塑料(例如聚苯乙烯、丙烯腈丁二烯苯乙烯、聚碳酸酯);玻璃;微孔过滤器(例如纤维素、尼龙、玻璃纤维、聚酯和聚碳酸酯);用于分批或连续细胞培养或基因工程(例如生物反应器)的生物反应器的材料,其可以包括中空纤维管或微载体珠;聚四氟乙烯陶瓷和相关聚合物材料。
[0196]
在特定方面,细胞培养物不含或基本上不含任何细胞外基质蛋白,例如层粘连蛋白、纤连蛋白、玻连蛋白、matrigel
tm
、腱生蛋白、巢蛋白、血小板反应蛋白、弹性蛋白、明胶、胶原蛋白、原纤维蛋白、分区蛋白、锚定蛋白、软骨粘连蛋白、连接蛋白、骨唾液蛋白、骨钙蛋
白、骨桥蛋白、epinectin、透明质蛋白、粗纤维调节素、表皮整联配体蛋白和缰蛋白。
[0197]
d.hpc向小胶质细胞的分化
[0198]
小胶质细胞是中枢神经系统的先天免疫细胞,其在大脑发育、体内平衡和免疫调节中发挥关键作用。它们很难从人胎儿和初生组织中获得。在某些实施方案中,本发明的方法描述了在确定的条件下从附加型重编程的hpc中产生、表征和冷冻保存人ipsc衍生的小胶质细胞(imgl)。冷冻保存的imgl保持纯度、分泌免疫调节细胞因子和吞噬phrodo red标记的细菌生物颗粒和淀粉样蛋白β聚集体。产生基本上无限数量的imgl的能力为加速对小胶质细胞在正常和疾病状态中的作用的人神经科学研究提供了巨大的希望。
[0199]
在示例性方法中,将新鲜或冷冻保存的hpc解冻并铺板在包含flt-3配体和il-3的小胶质细胞分化培养基中。细胞可以以10-50k/cm2例如20-35k/cm2的密度铺板。小胶质细胞分化培养基可包含il-34、tgfβ1或m-csf(mdm)。培养可以在matrigel
tm
包被的板或带电表面(例如primaria板或超低附着板或组织培养板(tc)或非组织培养板(non-tc))上进行,并且可以是高通量的,例如96孔板(例如,每孔200μl小胶质细胞分化培养基)。在接下来的23天分化过程中,细胞可以每48小时用每孔50μl的2x小胶质细胞分化培养基(mdm)进行半补料。在具体方面,分化是在不存在ecm蛋白(例如)的情况下进行的。在第23天用冷pbs收集细胞,并使用自动细胞计数仪定量总活细胞数。针对cd11b、cd11c、cd45、cd33、trem-2的表面表达和trem-2、iba、cx3cr1、p2ry12和tmem119的细胞内表达对细胞进行染色。
[0200]
e.内皮细胞
[0201]
在e8的存在下维持在matrigel
tm
或玻连蛋白上的ipsc可以适应缺氧至少5-10代。细胞可以从亚汇合ipsc中分开,并在存在补充有5um blebbistatin或1um h1152的无血清(sfd)培养基的情况下以25万个细胞/孔的密度铺板到胺培养皿上。铺板后24小时补充有50ng/ml bmp4、vegf和fgf2的sfd培养基可以添加到培养物中。在整个分化过程中,细胞可以每48小时进行补料。整个过程可以在缺氧条件下进行。在分化结束时收获的细胞可以冷冻保存或以25k/cm2的密度重新铺在羧基表面上,以在存在vasculife vegf内皮培养基或sfd内皮培养基的情况下启动内皮分化。
[0202]
在示例性方法中,在存在vasculife vegf内皮培养基或sfd内皮培养基和缺氧条件的情况下,将冷冻保存的第6天hpc或活培养物以25k/cm2铺板在羧基表面上。铺板后24小时给细胞提供内皮培养基的新鲜进料,并且培养物每48小时进行补料直到它们达到汇合。细胞可能需要5-6天才能达到汇合。使用tryple select收获细胞,针对表面内皮标志物cd31、cd105和cd144进行染色,然后用内皮培养基以25k/cm2重新铺板到羧基表面,并置于缺氧培养箱条件中。在分开后的第2、4、6天,给细胞提供内皮培养基的完整进料。在第7天,收获细胞、染色并以同样的方式重新铺板另外3次。
[0203]
在一些实施方案中,内皮细胞被转化为脑微血管内皮细胞。在示例性方法中,将活的或冷冻保存的hpc(例如,在补充有bmp4、vegf和fgf2的sfd的存在下在胺表面上衍生的第7天hpc)铺板到含有纤连蛋白(例如,50-200μg/ml,特别是100μg/ml)和胶原蛋白i(例如,100-500μg/ml,特别是400μg/ml)与ecra培养基(人内皮sfm[gibco],1%血小板贫乏血浆来源的牛血清[fisher],20ng/ml bfgf[promega],10um视黄酸)的ecm上。细胞可以以50-100k/cm2、特别是75k/cm2的密度铺板。培养物可以在缺氧培养箱条件下维持。培养物可以每
隔一天补料ecra培养基直至汇合。然后收获汇合的培养物,例如通过使用tryple。可以对收获的细胞进行染色以检测pecam-1(cd31)和glut-1。收获的细胞可以重新铺板,例如在具有ecra培养基的transwell插入物上,并放置在缺氧培养箱条件中。培养物可以每隔一天补料ecra培养基直至汇合。可以测试汇合培养物的跨内皮电阻(teer)。
[0204]
f.间充质细胞
[0205]
在一些实施方案中,ipsc分化为msc。例如,图5c显示了生成msc的2d hpc分化过程的示意图。将在e8存在的情况下在matrigel
tm
或玻连蛋白上维持的ipsc适应缺氧至少5-10代。将细胞从亚汇合ipsc中分开,并以25万个细胞/孔的密度在存在补充有5um blebbistatin或10um h1152的无血清的成分确定的(sfd)培养基的情况下铺板到胺培养皿上。铺板后24小时,将补充有50ng/ml bmp4、vegf和fgf2的sfd培养基添加到培养物中。在整个分化过程中,细胞每48小时进行补料。整个过程在缺氧条件下进行。在分化的第6天或第7天结束时,将细胞置于gmp
–
msc培养基中。允许细胞生长至汇合并在每次传代结束时收获,然后以50k/cm2的密度重新铺板在补充有5um blebbistatin或10um h1152的gmp-msc培养基中的胺表面上,以选择性地允许msc的生长和增殖。
[0206]
在一些方面,将冷冻保存的第6天hpc或第6天分化结束时的活培养物在msc培养基和10um h1152的存在下置于胺带电表面平板上。铺板后24小时给细胞提供msc培养基的新鲜进料,并且培养物每48小时进行补料直到它们达到汇合。细胞达到汇合需要5-6天。使用tryple收集细胞,并针对表面msc标志物cd73、cd44、cd105、cd49d以及内皮标志物cd31和cd144的不存在进行染色。在缺氧条件和胺表面下,使用上述过程将出现的培养物传代三次。将培养物在p4时转移至常氧和正常组织培养板。
[0207]
在一些方面,msc可以进一步分化为周细胞。在示例性方法中,将msc接种到sciencell周细胞培养基(目录号:1201)中(例如,在组织培养塑料(tcp)6孔板上以1-20k/cm2的细胞密度,特别是10k/cm2的细胞密度)并置于常氧培养箱条件中。培养物可以每隔一天补料sciencell周细胞培养基直至汇合。可以收获汇合的培养物,例如通过使用tryple。可以对收获的细胞进行染色以检测神经胶质抗原2/硫酸软骨素蛋白多糖(ng2)和pdgfr-β(cd140b)。收获的细胞可以在sciencell周细胞培养基中重新铺板(例如,在tcp 6孔板上以1-20k/cm2的细胞密度,特别是10k/cm2的细胞密度)。培养物可以每隔一天补料sciencell周细胞培养基直至汇合,然后以与上述相同的方式收获和染色。然后将细胞重新铺板,直到培养物扩增并保持纯度。此外,细胞可以针对cd146、cd49a、cd166、cd54、cd73、cd105、cd13、cd56、cd49d和/或cd44的存在阳性染色。
[0208]
g.分化培养基
[0209]
可以用支持每个具体细胞群的生长所必需的营养物来培养细胞。通常,细胞在包含碳源、氮源和维持ph的缓冲液的生长培养基中培养。培养基还可含有脂肪酸或脂质、氨基酸(例如非必需氨基酸)、维生素、生长因子、细胞因子、抗氧化物质、丙酮酸、缓冲剂、ph指示剂和无机盐。示例性生长培养基包含基本必需培养基,例如杜氏改良伊格尔培养基(dmem)或essential 8
tm
(e8
tm
)培养基,其补充有各种营养物,例如非必需氨基酸和维生素,以促进干细胞生长。基本必需培养基的实例包括但不限于基本必需培养基伊格尔(mem)α培养基、杜氏改良伊格尔培养基(dmem)、rpmi-1640培养基、199培养基和f12培养基。此外,基本必需培养基可以补充添加剂,例如马、小牛或胎牛血清。或者,培养基可以是无血清的。在其他情
况下,生长培养基可包含“敲除血清替代物”,其在本文中被称为被优化以在培养物中生长和维持未分化细胞例如干细胞的无血清制剂。knockout
tm
血清替代物公开于例如美国专利申请号2002/0076747中,其通过引用并入本文。优选地,psc在完全确定且无饲养层的培养基中培养。
[0210]
在一些实施方案中,培养基可以含有或不含血清的任何替代物。血清的替代物可以包括适当地含有白蛋白的材料(例如富含脂质的白蛋白、白蛋白替代物例如重组白蛋白、植物淀粉、葡聚糖和蛋白质水解物)、转铁蛋白(或其他铁转运蛋白)、脂肪酸、胰岛素、胶原蛋白前体、微量元素、2-巯基乙醇、3'-硫代甘油或其等价物。例如,可以通过国际公开号wo98/30679中公开的方法制备血清的替代物。或者,为了更方便,可以使用任何市售材料。市售材料包括knockout
tm
血清替代物(ksr)、化学成分确定的脂质浓缩物(gibco)和glutamax
tm
(gibco)。
[0211]
可以适当地定义其他培养条件。例如,培养温度可为约30至40℃,例如至少或约31、32、33、34、35、36、37、38、39℃,但不特别限于此。在一个实施方案中,细胞在37℃培养。co2浓度可为约1至10%,例如约2至5%,或其中可推导出的任何范围。氧张力可以是至少、多至或约1、2、3、4、5、6、7、8、9、10、20%或其中可推导出的任何范围。
[0212]
h.冷冻保存
[0213]
通过本文公开的方法产生的细胞可以在该过程的任何阶段例如阶段i、阶段ii或阶段iii冷冻保存,参见例如pct公开号2012/149484a2,其通过引用并入本文。细胞可以在有或没有基底的情况下冷冻保存。在几个实施方案中,储存温度范围为约-50℃至约-60℃、约-60℃至约-70℃、约-70℃至约-80℃、约-80℃至约-90℃、约-90℃至约-100℃及其重叠范围。在一些实施方案中,较低的温度用于冷冻保存的细胞的储存(例如,维持)。在几个实施方案中,使用液氮(或其他类似的液体冷却剂)来储存细胞。在进一步的实施方案中,细胞储存大于约6小时。在另外的实施方案中,细胞储存约72小时。在几个实施方案中,细胞储存48小时至约一周。在其他实施方案中,细胞储存约1、2、3、4、5、6、7或8周。在进一步的实施方案中,细胞储存1、2、3、4、5、6、7、8、9、10、11或12个月。细胞也可以储存更长时间。细胞可以单独冷冻保存或在基底(例如本文公开的任何基底)上冷冻保存。
[0214]
在一些实施方案中,可以使用额外的冷冻保护剂。例如,可以将细胞冷冻保存在包含一种或多种冷冻保护剂例如dm80、血清白蛋白(例如人或牛血清白蛋白)的冷冻保存溶液中。在某些实施方案中,溶液包含约1%、约1.5%、约2%、约2.5%、约3%、约4%、约5%、约6%、约7%、约8%、约9%或约10%dmso。在其他实施方案中,溶液包含约1%至约3%、约2%至约4%、约3%至约5%、约4%至约6%、约5%至约7%、约6%至约8%、约7%至约9%或约8%至约10%二甲基亚砜(dmso)或白蛋白。在一个具体实施方案中,溶液包含2.5%dmso。在另一个具体实施方案中,溶液包含10%dmso。
[0215]
细胞可以在冷冻保存期间被冷却,例如以约1℃/分钟被冷却。在一些实施方案中,冷冻保存温度为约-80℃至约-180℃,或约-125℃至约-140℃。在一些实施方案中,细胞在以约1℃/分钟冷却之前冷却至4℃。冷冻保存的细胞可以在解冻用于使用之前转移到液氮的气相中。在一些实施方案中,例如,一旦细胞达到约-80℃,它们就被转移到液氮储存区。冷冻保存也可以使用速率受控冷冻机来完成。冷冻保存的细胞可以解冻,例如在约25℃至约40℃的温度下,并且通常在约37℃的温度下解冻。
[0216]
iii.使用方法
[0217]
本公开内容提供了一种可以产生大量多谱系细胞的方法。这些细胞群可用于许多重要的研究、开发和商业目的。这些包括但不限于细胞的体内移植或植入;在体外筛选抗病毒药物、细胞毒性化合物、致癌物、诱变剂、生长/调节因子、药物化合物等;阐明肝脏疾病和感染的机制;研究药物和/或生长因子的作用机制;诊断和监测患者的癌症;基因治疗;以及生物活性产品的生产,仅举几例。
[0218]
a.药物组合物
[0219]
本文还提供了包含本发明的细胞和药学上可接受的载体的药物组合物和制剂。
[0220]
根据本发明用于施用于受试者的细胞组合物因此可以使用一种或多种生理学可接受的载体以任何常规方式配制,所述载体包括促进将化合物加工成可药用制剂的赋形剂和助剂。适当的制剂取决于所选择的施用途径。
[0221]
如本文所述的药物组合物和制剂可以通过将具有所需纯度的活性成分(例如细胞)与一种或多种任选的药学上可接受的载体(remington's pharmaceutical sciences 22
nd edition,2012)混合以冻干制剂或水溶液的形式制备。药学上可接受的载体在所采用的剂量和浓度下对接受者通常是无毒的,并且包括但不限于:缓冲剂,例如磷酸盐、柠檬酸盐和其他有机酸;抗氧化剂,包括抗坏血酸和蛋氨酸;防腐剂(例如十八烷基二甲基苄基氯化铵;氯化六甲铵;苯扎氯铵;苄索氯铵;苯酚、丁醇或苯甲醇;对羟基苯甲酸烷基酯例如对羟基苯甲酸甲酯或丙酯;邻苯二酚;间苯二酚;环己醇;3-戊醇;和间甲酚);低分子量(少于约10个残基)多肽;蛋白质,例如血清白蛋白、明胶或免疫球蛋白;亲水性聚合物,例如聚乙烯吡咯烷酮;氨基酸,例如甘氨酸、谷氨酰胺、天冬酰胺、组氨酸、精氨酸或赖氨酸;单糖、二糖和其他碳水化合物,包括葡萄糖、甘露糖或糊精;螯合剂例如edta;糖例如蔗糖、甘露糖醇、海藻糖或山梨糖醇;形成盐的反离子,如钠;金属配合物(例如锌-蛋白质配合物);和/或非离子表面活性剂,例如聚乙二醇(peg)。本文的示例性药学上可接受的载体进一步包括间质药物分散剂,例如可溶性中性活性透明质酸酶糖蛋白(shasegp),例如人可溶性ph-20透明质酸酶糖蛋白,例如rhuph20(baxter international,inc.)。某些示例性shasegp和使用方法,包括rhuph20,在美国专利公开号2005/0260186和2006/0104968中有所描述。在一个方面,shasegp与一种或多种额外的糖胺聚糖酶例如软骨素酶组合。
[0222]
b.用于商业、治疗和研究目的的分销
[0223]
在一些实施方案中,提供了一种试剂系统,其包括在制造、分销或使用期间的任何时间存在的细胞。试剂盒可以包含本公开内容中描述的细胞与未分化的多能干细胞或其他分化的细胞类型(通常共有相同的基因组)的任何组合。每种细胞类型可以在同一实体或共享业务关系的不同实体的控制下,在同一设施中或在不同地点、在相同或不同时间被包装在一起,或包装在单独的容器中。药物组合物可以任选地包装在合适的容器中,并带有用于所需目的(例如机制毒理学)的书面说明。
[0224]
在一些实施方案中,提供了可包括例如一种或多种用于产生细胞的培养基和组分的试剂盒。在合适的情况下,试剂系统可以以水性介质或冻干形式包装。试剂盒的容器装置通常包括至少一个小瓶、试管、烧瓶、瓶子、注射器或其他容器装置,可将组分放入其中并且优选地适当等分。当试剂盒中有不止一种组分时,该试剂盒通常还将包含第二、第三或其他额外容器,额外的组分可以单独放置在其中。然而,小瓶中可以包含各种组分的组合。试剂
盒的组分可以作为干燥粉末提供。当试剂和/或组分以干粉形式提供时,可以通过添加合适的溶剂来复原粉末。考虑了溶剂也可以提供在另一种容器装置中。本公开内容的试剂盒还将通常包括用于在密闭空间中容纳试剂盒组分以用于商业销售的装置。这种容器可以包括注射或吹塑塑料容器,所需的小瓶保持在其中。该试剂盒还可包括使用说明,例如印刷或电子格式,例如数字格式。
iv.实施例
[0225]
包括以下实施例以进料说明本发明的优选实施方案。本领域技术人员应当理解,以下实施例中公开的技术代表了发明人发现的在本发明的实践中发挥良好作用的技术,因此可以认为构成了其实践的优选模式。然而,本领域技术人员根据本公开内容应当理解,在不脱离本发明的精神和范围的情况下,可以在所公开的具体实施方案中进行许多改变并且仍然获得相似或类似的结果。
[0226]
实施例1
–
内皮细胞的产生
[0227]
使在e8的存在下维持在matrigel
tm
或玻连蛋白上的ipsc适应缺氧至少5-10代。将细胞从亚汇合ipsc中分开,并以25万个细胞/孔的密度在补充有5um blebbistatin或1um h1152的无血清的成分确定(sfd)培养基的存在下铺板到胺培养皿上。铺板后24小时,将补充有50ng/ml bmp4、vegf和fgf2的sfd培养基添加到培养物中。在整个分化过程中,细胞每48小时进行补料。整个过程在缺氧条件下进行。在分化结束时收获细胞,并且可以将其冷冻保存或在存在vasculife vegf内皮培养基或sfd内皮培养基的情况下以25k/cm2的密度重新铺板在羧基表面上以启动内皮分化(图2)。
[0228]
在存在1μm h1152和缺氧条件的情况下,在存在vasculife vegf内皮培养基的情况下,将冷冻保存的第6天hpc或活培养物以25k/cm2铺板在羧基表面上。铺板后24小时给细胞提供vasculife的新鲜进料,并且培养物每48小时进行补料,直到它们达到汇合。细胞达到汇合需要5-6天。使用accumax在最小搅拌或移液下收获细胞,针对表面内皮标志物cd31、cd105和cd144进行染色,并用vasculife h1152以25k/cm2重新铺板到羧基表面上,并置于缺氧培养箱条件中。在分离后的第2、4、6天,给细胞提供vasculife的完整进料。在第7天,收获细胞、染色并以同样的方式重新铺板另外3次。直方图描绘了在每个重新铺板阶段内皮细胞纯度的增加。使用连续传代纯化而不使用cd31 macs产生纯的内皮细胞。内皮细胞可以在重新铺板第3代结束时进行冷冻保存(图3)。
[0229]
实施例2
–
间充质干细胞的产生
[0230]
图5c显示了生成msc的2d hpc分化过程的示意图。使在e8的存在下维持在matrigel
tm
或玻连蛋白上的ipsc适应缺氧至少5-10代。将细胞从亚汇合ipsc中分开,并以25万个细胞/孔的密度在补充有5um blebbistatin或1um h1152的无血清的成分确定(sfd)培养基的存在下铺板到胺培养皿上。铺板后24小时,向培养物中加入补充有50ng/ml bmp4、vegf和fgf2的sfd培养基。在整个分化过程中,细胞每48小时进行补料。整个过程在缺氧条件下进行。在分化的第6/7天结束时,将细胞置于gmp-msc培养基中。前体群体的表型在收获后进行了分析(图5c)。使细胞生长至汇合并在每次传代结束时收获,然后以50k/cm2的密度重新铺板在补充有5um blebbistatin或1um h1152的gmp-msc培养基中的胺表面上,以选择性地允许msc的生长和增殖。
[0231]
将来自3d/2d hpc分化的冷冻保存的第6天hpc或来自2dhpc分化的第6天分化结束时的活培养物在msc培养基和1um h1152的存在下置于胺带电表面平板上。铺板后24小时给细胞提供msc培养基的新鲜进料,并且培养物每48小时进行补料,直到它们达到汇合。细胞达到汇合需要5-6天。使用tryple收集细胞,并针对表面msc标志物cd73、cd44、cd105、cd49d以及内皮标志物cd31和cd144的缺失进行染色。在缺氧条件和胺表面下,使用上述过程将出现的培养物传代三次。在p4时将培养物转移至常氧和正常组织培养板。msc的纯度规格在p6处达到(图6、7)。将在p3处冷冻保存的msc解冻并放入如图8a中所述的谱系特异性分化基质中,以证明产生骨细胞、软骨细胞和脂肪细胞的三系潜能(图8b)。通过在10cm组织培养板中以1000个细胞/cm2的密度铺板msc,证明了冷冻保存的msc的克隆增殖能力。细胞用msc培养基进行补料,持续2周,其中每隔一天更换培养基。出现的集落用结晶紫染色并评分(图8c)。
[0232]
实施例3
–
从msc产生周细胞
[0233]
icell msc和ipsc衍生的周细胞在50%汇合时进行取样并针对已知周细胞标志物pdgfrβ、ng2和cd146通过流式细胞术进行分析。将冷冻保存的msc解冻并以35,000个细胞/cm2在没有细胞外基质(ecm)的情况下在msc维持培养基中铺板在6孔板中(图9a)。使细胞达到汇合并且将细胞以15,000个细胞/cm2在没有细胞外基质(ecm)的情况下在sfd周细胞培养基(spm)中重新铺板在6孔板中(图9a;图9b)。
[0234]
将原代人脑血管周细胞(hbvp)(sciencell#1200)解冻并以5,000个细胞/cm2在周细胞培养基(sciencell#1201)中铺板在聚-l-鸟氨酸包被的6孔板上。这些细胞在分化过程中用作阳性对照。sciencell hbvp、icell msc和ipsc衍生的周细胞针对已知周细胞标志物pdgfrβ、ng2和cd146通过流式细胞术进行分析(图9c)。解冻时不存在icell msc的周细胞标志物。hbvp和ipsc衍生的周细胞显示已知周细胞标志物pdgfrβ、ng2和cd146的表达,其中ipsc衍生的周细胞具有比sciencell hbvp更高的纯度(图9c)。ipsc衍生的周细胞表现出与sciencell hbvp相似的形态(图9d)。
[0235]
基于它们的功能,周细胞可以被分类为表型pc1(促炎)或pc2(收缩)(rustenhoven等人,2017)。两种亚型的特征在图9e中描述。解冻后,ipsc衍生的周细胞针对pc1和pc2标志物cd274、vcam1、钙调蛋白、结蛋白、dlk1和αsma通过流式细胞术进行亚分型(图9f)。ipsc衍生的周细胞揭示了收缩周细胞(pc2亚型)的特征。
[0236]
除了在慢性和急性bbb模型中看到的非特异性吞噬细胞摄取外,周细胞还通过在生理和病理条件下处理某些大分子的清除来特异性地调节它们的神经元微环境(winkler等人,2014)。将ipsc衍生的周细胞以15,000个细胞/cm2在spm中铺板在具有pdl涂层(greiner#655946)的96孔板中。在将死亡指示剂nucgreen dead 488(invitrogen#r37109)和金黄色葡萄球菌phrodo red生物颗粒(invitrogen#a10010)加入细胞之前,使细胞在铺板后静息三天。将板放置在incucyte实时成像系统上持续一个多月,每周补料一次(包括相同浓度的活/死和生物颗粒试剂)。ipsc衍生的周细胞显示出高于对照的金黄色葡萄球菌生物颗粒的可观察到的吞噬活性。
[0237]
实施例4-脑微血管内皮细胞(bmec)的产生
[0238]
使在e8的存在下维持在matrigel
tm
或玻连蛋白上的ipsc适应缺氧至少5-10代以产生脑微血管内皮细胞。将活的或冷冻保存的hpc(例如,在补充有bmp4、vegf和/或fgf2例如bmp4和fgf2的sfd的存在下,在胺表面衍生的第6天hpc)铺板到含有纤连蛋白(例如,50-200
μg/ml,特别是100μg/ml)和胶原iv(例如,100-500μg/ml,特别是400μg/ml)与ecra培养基(人内皮sfm(gibco),1%血小板贫乏的血浆来源的牛血清(fisher)、20ng/ml bfgf(promega)、10um视黄酸)的ecm上。细胞以50-100k/cm2、特别是75k/cm2的密度铺板。培养物每天补料培养并维持在缺氧培养箱条件下。培养物每隔一天补料ecra培养基直至汇合。然后收获汇合的培养物,例如通过使用tryple。对收获的细胞进行染色以检测pecam-1(cd31)和glut-1以确认bmec的身份(图10b)。将收获的细胞重新铺板,例如在带有ecra培养基的transwell插入物上,并放置在缺氧培养箱条件中。(图10a)。培养物可以每隔一天补料ecra培养基直至汇合。可以通过流式细胞术(图10c)和免疫细胞化学(图10d)和跨内皮电阻(teer)测试汇合的培养物中p-gp、cd105、glu-1和cd31表达的存在,并与空白培养基进行比较(图10e)。对于免疫组织化学,细胞用200μl dpbs洗涤3次,然后与兔抗p-gp抗体(在封闭缓冲液(dpbs中的10%fbs,0.01%tritonx)中的1:50)在4℃下孵育过夜。用200μl dpbs洗涤3次后,p-gp用二抗(1:1000,驴抗兔igg alexa fluor 488(invitrogen))染色。细胞核用hoechst3342(thermo fisher)染色。图像由imagexpress(molecular devices,llc)以200倍放大率捕获。
[0239]
实施例5-小胶质细胞的产生
[0240]
使在e8的存在下维持在matrigel
tm
或玻连蛋白上的ipsc适应缺氧至少5-10代。2d hpc分化:将细胞从亚汇合ipsc中分开,并在补充有5um blebbistatin或1um h1152的无血清的成分确定(sfd)培养基中以25-50万个细胞/孔的密度铺板到胺培养皿上。铺板后24小时,将补充有50ng/ml bmp4、vegf和fgf2的sfd培养基添加到培养物中。第二天进行完全培养基交换。
[0241]
在分化过程的第五天,将细胞置于含有50ng/ml flt-3配体、scf、tp0、il3和il6和5u/ml肝素的培养基中。在整个分化过程中,细胞每48小时进行补料。整个过程在缺氧条件下进行。hpc通过cd43/cd34细胞的存在进行定量。
[0242]
3d hpc分化:细胞从亚汇合的ipsc分开,并在补充有5um blebbistatin或1um h1152的无血清的成分确定(sfd)培养基的存在下以25-50万个细胞/ml的密度铺板到旋转瓶中。铺板后24小时更换补充有50ng/ml bmp4、vegf和fgf2的sfd培养基。在分化过程的第五天,将细胞置于含有50ng/ml flt-3配体、scf、tp0、il3和il6以及5u/ml肝素的培养基中。在整个分化过程中细胞每48小时进行补料。整个过程在缺氧条件下进行。hpc通过cd43/cd34的存在进行定量。过程概述和效率在(图11)中举例说明并且培养基组成在(图12)中列出。
[0243]
将hpc置于小胶质细胞分化培养基mdm或2x-mdm中(图11)。培养物每48小时进行补料。在冷冻保存之前和之后对分化第23天的小胶质细胞培养物的纯度标志物进行定量(图13a、图13b)。
[0244]
评估第23天活的和冷冻保存的小胶质细胞培养物的纯度。收获第23天分化时的小胶质细胞培养物,并针对小胶质细胞特异性标志物的存在进行染色。使用速率受控冷冻机冷冻保存剩余的细胞。将冷冻保存的细胞解冻并针对小胶质细胞特异性标志物的存在进行染色。对于这两组,cd45、cd33、trem2和cd11b的细胞表面表达(图14a)以及pu.1、iba、p2ry12、trem2和tmem119的细胞内表达通过流式细胞术确定(图14b)。结果表明,冷冻保存的小胶质细胞在冷冻保存后仍保持纯度。
[0245]
将hpc在mdm的存在下置于培养基中以启动小胶质细胞分化,并用2x-mdm进行间歇进料。在分化的第20、23和26天使用手动冷冻方案或速率受控冷冻机(crf)冷冻保存细胞。将冷冻保存的细胞转移到液氮中一周。将冷冻保存的小胶质细胞解冻并置于小胶质细胞成熟培养基(mmm)中。培养物每48小时用新鲜的小胶质细胞成熟培养基进行补料。在解冻后第3、5、7、10、12和14天收获细胞,并定量相对于初始铺板数的活细胞回收率(图15a-15c)。
[0246]
在mdm的存在下,将冷冻保存的hpc分化为小胶质细胞。定量输入hpc和输出小胶质细胞的总存活数。基于小胶质细胞分化第23天存在的trem2阳性细胞的纯度和绝对数量除以输入活hpc的绝对数量来计算过程效率(图16)。
[0247]
在分化的第20天(图17a)、第23天(图17b)或第26天(图17c)冷冻保存的小胶质细胞在小胶质细胞成熟培养基(mmm)中解冻并每48小时补料新鲜培养基。在解冻后第3、7和10天定量总活力和绝对细胞数。数据显示第23天的小胶质细胞比第26天的小胶质细胞有更高的解冻后恢复率(图17a-17c)。
[0248]
接下来,在分化过程的第20天、第23天和第26天对冷冻保存的小胶质细胞进行功能评估。在每孔存在200μl小胶质细胞成熟培养基的情况下,将细胞解冻并以15,000个活细胞/孔铺板在96孔板中。用稀释的1μg/孔调理或非调理phrodo red生物颗粒(thermo fisher#a10010,每瓶2mg;储存在-20℃)处理细胞。将板置于incucyte上,并在解冻后最多5天的不同时间点拍摄吞噬作用的图像。通过速率受控冷冻方法冷冻保存的细胞表现出更强的吞噬作用(由于更高的细胞活力(图19))。
[0249]
功能评估扩展到解冻后的较晚时间点。对于使用手动或速率受控冷冻机在分化的第20、23和26天冷冻保存的小胶质细胞通过在incucyte系统上实时成像评估在解冻后第5、7和14天的吞噬潜能。将冷冻保存的小胶质细胞解冻并铺板在mmm中三天。三天结束时的活细胞计数如图18b中所述确定。将15,000个活细胞在存在每孔200μl小胶质细胞成熟培养基(mmm)以及稀释的1μg/孔调理或非调理phrodo red生物颗粒的情况下铺板在96孔板中,然后将板放置在incucyte上并在解冻后多至5、7和14天的不同时间点获得吞噬作用的图像。手动冷冻保存方法显示在所有条件下吞噬作用的降低/右移的速率(由于细胞活力降低)。
[0250]
吞噬指数是吞噬活性的量度,通过计算在细菌和吞噬细胞的悬浮液的有限孵育期间每个吞噬细胞摄取的细菌数来确定。冷冻保存的小胶质细胞吞噬标记的细菌颗粒的能力通过吞噬红色物体计数/总活细胞数的比率来定量。该比率被确定为吞噬指数(图21)。
[0251]
将冷冻保存的小胶质细胞解冻并在存在每孔200μl小胶质细胞成熟培养基的情况下以15,000个活细胞/孔铺板在96孔板中。用稀释的1μg/孔调理或非调理phrodo red生物颗粒(thermo fisher#a10010,每瓶2mg;储存在-20℃)处理细胞。将板置于incucyte上,并在解冻后最多5天的不同时间点拍摄吞噬作用的图像。通过速率受控冷冻方法冷冻保存的细胞表现出更强大的吞噬作用(由于更高的细胞活力(图22))。
[0252]
接下来,在不存在ecm的情况下并以适合筛选应用的96孔格式进一步开发了hpc向小胶质细胞的分化。在超低附着(ula)、组织培养(tc)和非组织培养(non-tc)容器上进行的分化(图23a)。在存在每孔200ul小胶质细胞分化培养基的情况下,将冷冻保存的hpc以20,000-35,000个活细胞/cm2的密度铺板在96孔primaria板或超低附着、组织培养(tc)或非组织培养板(non-tc)上每孔培养基(图23b)。在接下来的23天分化中,每48小时向细胞中每孔mdm加入50μl培养基。在第23天用冷pbs收集细胞,并使用自动细胞计数器定量总活细胞数。
针对cd11b、cd45、cd33、trem2的表面表达和trem2、iba、p2ry12和tmem119的细胞内表达对细胞进行染色(图24a-24b)。
[0253]
表1:在带电表面上产生小胶质细胞的过程效率。
[0254]
板类型第0天细胞数第23天细胞数扩增primera0.684
×
1064.0
×
1065.8xula0.684
×
1062.06
×
1063xtc0.684
×
1063.22
×
1064.7xnon-tc0.684
×
1063.52
×
1065.1x
[0255]
由冷冻保存的小胶质细胞释放的细胞因子和趋化因子。将第23天冷冻保存的小胶质细胞解冻到mdm培养基中并以50,000个细胞/孔铺板到primaria 96孔板上。在开始用100ng/ml lps和50ng/ml干扰素γ刺激之前,将细胞铺板三天。刺激以一式三份进行,持续24小时。将上清液离心以去除细胞和碎片,并立即置于-20℃。在多重luminex测定上分析上清液。
[0256]
实施例6-工程化ipsc以产生模拟神经变性疾病的变体
[0257]
通过在外显子2中引入插入缺失来破坏trem2功能,导致移码和翻译提前终止。tal核酸酶(配对下面的trem2)被设计为结合以外显子2内的氨基酸58为中心的dna序列。用于工程化的细胞系是fcdi ipsc系01279.107。使用设置为125v/950uf的biorad gene pulser xcell系统将tal核酸酶mrna和在sv40启动子控制下表达杀稻瘟素抗性的共选择质粒电穿孔到细胞中。将细胞铺板并在电穿孔后的第1天和第2天应用短的杀稻瘟素选择。生长存活细胞,然后在电穿孔后第7天将单细胞分选到96孔板中。大约两周后,挑选出81个克隆并通过pcr和测序进行基因分型。
[0258]
在测序的81个克隆中,7个显示出序列修饰。3个克隆含有具有1个碱基对插入的1个等位基因,3个克隆含有具有1个碱基对缺失的1个等位基因,1个克隆为其中1个等位基因含有1个碱基对插入和1个等位基因4个碱基对缺失的复合杂合子。第七个克隆包含预计不会引入移码的24个碱基对的缺失。克隆被扩增、冷冻保存,并进行序列确认和核型分析。分化为小胶质细胞后,选择两个主要克隆作为杂合或纯合破坏的实例。杂合克隆01279.1185包含具有1bp插入的等位基因,导致在trem2的第60位发生移码并在45个后续氨基酸后终止。纯合克隆01279.1187含有在第59位具有1bp移码插入并在16个氨基酸后终止的等位基因,和在第59位具有4bp缺失从而导致移码并在46个氨基酸后终止的第二个等位基因。
[0259]
表2:靶序列。
[0260]
tal靶序列trem2-fggtgccgccagctgggagtrem2-rgcgtgctgaccacacgct
[0261]
表3
[0262][0263]
实施例7-模拟神经变性的额外等基因工程化系的产生:
[0264]
帕金森病模型是通过核酸酶介导的同源重组和供体寡核苷酸sjd 14-133通过基因工程化附加体重编程的ipsc 01279产生的。所得ipsc含有snp rs104893877,其中氨基酸53从丙氨酸变为苏氨酸,导致α-突触核蛋白基因(snca)中的a53t变体,以及导致snca a53t ipsc系的两个沉默突变。
[0265]
通过使用核酸酶介导的同源重组和供体质粒p1553产生用于研究雷特综合征的等基因工程化模型。供体质粒p1553在甲基cpg结合结构域之前插入一系列终止密码子,然后是侧接loxp位点的pgkp-puromycinr-sv40pa选择盒。源自亲本系01279的mecp2hm系为雷特综合征提供了疾病模型。
[0266]
从等基因工程化ipsc产生hpc和小胶质细胞:来自01279 ipsc的纯合和杂合trem2 ko ipsc以及衍生自01279的snca a53t和mecp2 hm工程化系在e8和matrigel
tm
的存在下维持,通过将它们传代10代来适应缺氧条件。对细胞进行核型分析,并通过3dhpc分化方案使ipsc库启动hpc分化。将细胞从亚汇合ipsc中分开,并在补充有5μm blebbistatin或1μm h1152的无血清的成分确定(sfd)培养基的存在下以每毫升25-50万个细胞的密度铺板到旋转瓶中。铺板后24小时更换补充有50ng/ml bmp4、vegf和fgf2的sfd培养基。在分化过程的第五天,将细胞置于含有50ng/ml flt-3配体、scf、tp0、il3和il6以及5-10u/ml肝素的培养基中。在整个13天分化过程中,细胞每48小时进行补料。整个过程在缺氧条件下进行。通过cd43/cd34的存在定量hpc。在使用cd34珠进行macs分选后冷冻保存hpc。小胶质细胞是通过解冻冷冻保存的hpc并将细胞置于如实施例5中所述的23天分化过程中产生的。
[0267]
来自第23天野生型和trem工程化克隆的冷冻保存的小胶质细胞被解冻,并且trem-2表达连同cd45的存在通过流式细胞术定量(图26)。
[0268]
来源于等基因工程化系的冷冻保存的第23天的小胶质细胞被解冻并针对小胶质细胞特异性标志物的存在染色。通过流式细胞术对细胞进行染色以定量cd45、cd33、trem2和cd11b的细胞表面表达以及pu.1、iba、p2ry12、trem2和tmem119蛋白的细胞内表达。图26总结了所有四种等基因工程化ipsc中获得的纯度。结果表明,在没有改变分化方案的情况下,从等基因工程化的ipsc产生高纯度的小胶质细胞。
[0269]
使用simple step elisa(abcam)从收集自wt和trem2杂合和纯合ko突变体的条件培养基中定量解冻后小胶质细胞分泌的可溶性trem2(strem2)蛋白的水平(图27a)。将wt和trem2 ko小胶质细胞解冻和在96孔primaria板中在成熟培养基中以相同的密度铺板。在解冻后第3天和解冻后第7天收集用过的培养基。在解冻后的第3天和第5天,培养物用新鲜成熟培养基进行半补料。数据揭示了wt、杂合trem2 ko和纯合ko小胶质细胞之间可溶性trem2水平的差异。该测定可用作区分wt和trem2工程化ipsc的功能测定。
[0270]
使用simple step elisa(abcam)从收集自wt和trem2杂合和纯合ko突变体、mecp2hm和snca-a53t收集的条件培养基中定量解冻后小胶质细胞分泌的可溶性trem2(strem2)蛋白的水平(图27a)。将wt和trem2ko小胶质细胞解冻和在96孔primaria板中在成熟培养基中以相同的密度铺板。在解冻后第3天和解冻后第7天收集用过的培养基。在解冻后的第3天和第5天,培养物用新鲜成熟培养基进行半补料。数据揭示了wt、杂合trem2 ko和纯合ko小胶质细胞之间可溶性trem2水平的差异。该测定可用作区分wt和trem2工程化ipsc的功能测定。a53t-snca小胶质细胞中可溶性trem2的释放受到损害,而mecp2hm小胶质细胞确实揭示了培养基中释放的strem水平的任何改变(图27b)。
[0271]
由等基因工程化的冷冻保存的小胶质细胞释放的细胞因子和趋化因子。将来源于wt、1185ht trem2 ko、1187ho trem2 ko a53t-snca和mecp2hm小胶质细胞的第23天冷冻保存的小胶质细胞解冻到mdm培养基中,并以50,000个细胞/孔铺板到primaria96孔板上。在开始用100ng/ml lps刺激之前,将细胞铺板三天以检查m1介导的反应。刺激一式三份进行,持续24小时。将上清液离心以去除细胞和碎片,并立即置于-20℃。在多重luminex测定上分析上清液。该多重luminex测定的结果被捕获为图27c中的热图。与anh对照相比,工程化系分泌更高水平的il-6。与anh相比,trem2 hz和trem2 ho和mecp2hm小胶质细胞释放更少的tnfα,但释放增加的il6水平。与ahn对照小胶质细胞相比,a53t-snca小胶质细胞释放出相似水平的il-6和tnfα。
[0272]
当用m1刺激物(lps)处理时,所有工程化系释放m2细胞因子il-10。与ahn对照小胶质细胞相比,mecp2hm小胶质细胞释放更少的il-10(图27e)。ahn和工程化小胶质细胞能够响应lps刺激释放ccl2/mcp-1、ccl20/mip-3α、ccl4/mip-1β、ccl5/rantes、cx3cl1/分形趋化因子、cxcl1/groα、cxcl10/ip-10、cxcl2/groβ、il-8/cxcl8。细胞因子释放水平存在一些固有差异。trem2ho揭示了最高水平的ccl4,其是阿尔茨海默病(ad)发作期间释放的关键分析物。mecp2hm、trem2hz和trem2ho小胶质细胞释放出更高水平的cxcl1/gro,这意味着试图招募辅助细胞类型粒细胞以帮助杀死微生物并在吞噬过程中触发炎症反应,mecphm小胶质细胞显示自发分泌il-8/cxc18。这种分析物在脑损伤中升高,并诱导促炎蛋白酶和mmp-2和mmp-9的表达。mecphm小胶质细胞分泌更高水平的il-6。这些结果表明mecphm已准备好(primed)用于促炎反应。工程化和ahn小胶质细胞在培养基中响应lps释放相似水平的pdl-1、cd40、flt-3和pdgfaa。
[0273]
在进行筛选实验之前,将冷冻保存的小胶质细胞在成熟培养基中解冻并使其恢复48小时(图36)。来自trem2 wt和trem2 hoko的5,000个小胶质细胞在384孔板的每孔中铺板在40ul培养基中,持续24小时。在第一组(板1)中,细胞用终浓度为1μm的化合物进行预处理。用化合物处理细胞后24小时,将phrodo标记的淀粉样蛋白-β以1μm的终浓度添加到板1中,并在incucytes3上捕获吞噬作用96小时(图37-39)。在第二组(板2)中,将细胞铺板24小时,然后用终浓度为1μg/ml的1ug/ml lps处理(图40-42)。暴露于lps后24小时和初始铺板后48小时,phrodo标记的淀粉样蛋白-β以1μm的最终浓度添加到板2。使用incucyte每小时对细胞成像一次,持续多至96小时。吞噬数据被捕获为总红色物体综合强度xμm2/图像。所有处理的最终体积保持不变。筛选的结果总结在图43中。
[0274]
为了理解在成熟培养基中解冻后小胶质细胞存活所需的细胞因子,计划了具有32种不同培养基配方的示意性矩阵(图28)。将wt、1185 ht trem2 ko、1187 ho trem2 ko小胶
质细胞以15,000个活细胞的密度置于96孔板中的250μl小胶质细胞基础培养基或mmm,或补充有成熟培养基中的单一细胞因子(图29)、两种细胞因子(图30)、三种细胞因子(图31)或四种细胞因子(图32)的小胶质细胞基础培养基。在incucyte系统上捕获细胞存活的动力学。将稀释至2滴/ml的nucgreen dead添加到含有具有各种培养基组分的细胞的所有孔中,以随时间捕获死细胞的数量。每8小时捕获图像,实验持续72小时而没有任何间歇性进料。nucgreen dead的强度定量了培养物中的死细胞。
[0275]
将wt、1185 ht trem2 ko、1187 ho trem2 ko小胶质细胞以15,000个活细胞的密度置于96孔板中的250μl小胶质细胞基础培养基(图33a)或mmm(图33b)、补充有il-34的小胶质细胞基础培养基(26c)、补充有il-34的小胶质细胞基础培养基(图33d)、补充有mcsf的小胶质细胞基础培养基(图33d)或仅补充有il-34(图33c)或mscf或il-34和mcsf的组合(图33e)的基础培养基。在incucyte系统上捕获细胞存活的动力学。将稀释至2滴/ml的nucgreen dead添加到含有具有各种培养基组分的细胞的所有孔中,以随时间捕获死细胞的数量。每8小时捕获图像,实验持续7天而没有任何间歇性进料。nucgreen dead的强度定量了培养物中的死细胞。
[0276]
在wt、1185 ht trem2 ko、1187 ho trem2 ko小胶质细胞上评估功能表征,在第23天用phrodo red标记的细菌生物颗粒和phrodo red淀粉样蛋白β冷冻保存。wt、1185 ht trem2 ko、1187 ho trem2 ko小胶质细胞在解冻后三天以15,000-30,000个活细胞/cm2的密度铺板在96孔板中的250μl mmm(图33a-b)或仅补充mscf(图33c-d)或il-34(图33e-f)或il-34和mcsf的组合(图33g-h)的mdm基质(aka小胶质细胞基础培养基)中。用稀释的1μg/孔调理或非调理phrodo生物颗粒(thermo fisher#a10010,每瓶2mg;储存在-20℃)(图33a、c、e、g)或phrodo淀粉样蛋白β(图33b、d、f、h)处理细胞。将板置于incucyte上,并在最多30小时的不同时间点拍摄吞噬作用的图像。wt以及工程化的小胶质细胞显示出解冻后的吞噬功能。wt、1185 ht trem2 ko、1187 ho trem2 ko小胶质细胞的吞噬作用的动力学和效率不同。
[0277]
在成熟培养基中在存在mmm或补充有两种关键(il-34、mscf)细胞因子的组合的小胶质细胞基础培养基的情况下,确定第23天冷冻保存的野生型(wt)小胶质细胞的解冻后纯度(图35)。通过收获细胞在解冻后第3、7和14天定量纯度,并在分化过程结束时通过收获细胞并通过流式细胞术针对标志物的细胞表面和细胞内染色对细胞进行染色来确定cd45、cd33、trem2、cd11b、cx3cr1、p2ry12、tmem119、iba的纯度。冷冻保存的小胶质细胞在补充有mscf和il-34的成熟培养基中保持活力和纯度。这种简化的培养基对于冷冻保存的小胶质细胞与神经元和星形胶质细胞的共培养应用以用于开发大脑类器官模型以研究与神经变性相关的许多snp和突变的贡献非常有价值。
[0278]
实施例8-从患者衍生的ipsc产生疾病相关的小胶质细胞
[0279]
最近的遗传研究表明,几种小胶质细胞富集的基因中的多态性与发展阿尔茨海默病(ad)、帕金森病(pd)和几种神经变性疾病的风险改变有关。从显示trem2、cd33和abca7中的突变连同apoe同种型的供体中产生了总结来自一组末期小胶质细胞的gwas研究的风险相关snp的列表。来自患者衍生的ipsc的冷冻保存的小胶质细胞提供了一种体外工具以创建更准确的模型来了解2d或3d类器官系统中人小胶质细胞、神经元、星形胶质细胞之间的复杂相互作用,并模拟神经变性疾病(mcquade等人,2019)。
[0280]
从附加型重编程的ahn和疾病特异性ipsc生成hpc:在收集用于朝向造血细胞和随后小胶质细胞的分化的源材料之前,使用e8/matrigel
tm
使从正常供体和疾病特异性供体生成的附加型重编程ipsc适应缺氧至少5-10代。表4中描述了一组ipsc的基因型。对来源于所有供体的ipsc进行核型分析,并通过如实施例5所述进行的通过3d hpc分化方案使ipsc库启动hpc分化。通过解冻冷冻保存的hpc并将细胞置于如实施例5中所述的23天分化过程中产生小胶质细胞。来源于不同供体的冷冻保存的第23天小胶质细胞被解冻并针对小胶质细胞特异性标志物的存在染色。通过流式细胞术针对cd45、cd33、trem2和cd11b的细胞表面表达以及pu.1、iba、p2ry12、trem2和tmem119蛋白的细胞内表达对细胞进行染色。表5总结了在所有ahn和疾病相关小胶质细胞(dam)中获得的纯度。结果表明,在分化方案没有变化的情况下,从一组健康和疾病特异性供体中产生了高纯度的小胶质细胞。
[0281]
表4:从一组表观健康正常(anh)和疾病相关小胶质细胞(dam)产生冷冻保存的小胶质细胞
[0282]
ꢀꢀꢀ
apoeapoeapoetrem2cd33表型基因型性别rs429358rs7412基因型rs7593268rs12459419ahnn/a雌性t/tc/c3/3c/ct/tahnn/a雌性t/tc/c3/3c/ct/tahnn/a雌性t/tc/c3/3c/ct/tahntrem2 r47h雌性t/cc/t3/3c/tc/cahnn/a雄性t/tc/c3/3c/cc/cahncd33雌性t/cc/t2/4c/ct/tadapoe 4/4雌性c/cc/c4/4c/cc/cadapoe 4/4雌性c/cc/c4/4c/cc/cadapoe 4/4雌性c/cc/c4/4c/cc/tadapoe 4/4雌性c/cc/c4/4c/ct/tahnabca7 g1527a雄性t/tc/c3/3c/cc/tadapoe 2/4雌性t/tc/c2/4c/cc/t
[0283]
表5.小胶质细胞表观健康正常(anh)和阿尔茨海默病(ad)供体样品的纯度的概述。
[0284][0285]
使用simple step elisa(abcam)从收集自从一组ipsc供体产生的小胶质细胞的条件培养基中定量解冻后小胶质细胞分泌的可溶性trem2(strem2)蛋白的水平(图45)。从表观健康的正常供体和疾病特异性供体产生的小胶质细胞被解冻并以相同的密度铺板在
96孔primaria板的成熟培养基中。在解冻后第3天和解冻后第7天收集用过的培养基。在解冻后的第3天和第5天,培养物用新鲜成熟培养基进行半补料。
[0286]
数据揭示了来源于供体的小胶质细胞的各种样品之间的可溶性trem2水平的差异,来源于表现出r47h基因型的供体的小胶质细胞揭示了在解冻后第3天的最高水平的可溶性trem水平并且它甚至在解冻后7天保持高水平。尽管该供体无症状,因此被归类为ahn,但ipsc衍生的小胶质细胞分泌高水平的strem。该数据与在阿尔茨海默病患者的脑脊液中观察到的高水平strem2一致,并与这种突变状态相关(cheng等人,2016)。该数据是使用ipsc衍生的小胶质细胞设计筛选神经变性疾病发作的预测性试剂盒的有力实例。具有表现出ad发作的apoe4/4/基因型的年轻供体与具有相同基因型的老年供体相比显示出高水平的可溶性trem。cd33或abca7基因中snp的存在似乎并未增强培养基上清中可溶性trem的释放。ahn供体12068在解冻后3天和7天显示出高水平的可溶性trem。
[0287]
神经炎症促成许多神经变性疾病的进展和发病机制。大脑中的常驻小胶质细胞和星形胶质细胞取决于刺激和微环境释放可以在大脑中发挥促炎和抗炎作用的细胞因子。促炎和抗炎特征谱之间的这种波动与ad和其他神经变性疾病的发作有关。为了定量疾病相关小胶质细胞(dam)释放的细胞因子和趋化因子的水平,将冷冻保存的ahn和dam小胶质细胞解冻到mdm培养基中,并以50,000个细胞/孔铺板到primaria 96孔板上。在开始用100ng/ml lps刺激以检查m1介导的反应或用10ng/ml il-4和10um dbu-camp刺激以触发m2特异性反应之前,将细胞铺板三天。刺激一式三份进行,持续24小时。将上清液离心以去除细胞和碎片,并立即置于-20℃。在多重luminex测定上分析上清液。该多重luminex测定的结果被捕获为图27c中的热图。
[0288]
趋化因子ccl1、ccl2、ccl3、ccl4、ccl8、ccl11、ccl13、ccl17、ccl18、ccl20、ccl22、ccl24用作化学引诱物并介导髓样细胞、粒细胞、淋巴样细胞或神经前体细胞募集到发炎区域中,增强吞噬反应并通常在阿尔茨海默病(ad)或多发性硬化症中上调。响应lps或dbu-camp,与ahn系相比,源自apoe e4/e4、ap0e e2/e4、trem2 r47h、abca7 g1527a和cd33(带有rs429358 snp)衍生的小胶质细胞的小胶质细胞释放出更高水平的所有这些分析物。倍数增加在各种小胶质细胞基因型之间从0.1倍到高达7倍不等。来自疾病相关小胶质细胞的此数据支持了证明ad脑样品中ccl2和ccl5表达增加的早期发现。westin等人已报道大脑和脑脊液(csf)中的ccl2表达是ad严重程度的可靠预测因子。
[0289]
apoe e4/e4、ap0e e2/e4、trem2 r47h、abca7 g1527a和cd33(带有rs429358 snp)衍生的小胶质细胞释放略微升高水平的可溶性cd163,其是与m2极化相关的炎症和炎性疾病的标志物。scd163的释放防止单核细胞过度活化并减少促炎细胞因子tnf-α、il-1β、il-6和il-8的分泌。使用几丁质酶-3也观察到了类似的趋势,它在神经炎症期间的组织重塑中发挥作用(melief等人,2012;minett等人,2016)。
[0290]
pd-l1及其受体pd-1引发调节t细胞活化、耐受性和免疫介导的组织损伤之间的平衡的抑制信号。响应于lps,与ahn衍生的小胶质细胞相比,apoe e4/e4衍生的小胶质细胞没有增加。与ahn系相比,trem2 r47h、apoe e2/e4和cd33(带有rs429358 snp)衍生的小胶质细胞显示出增加。响应于il-4 dbu-camp,apoe e4/e4、trem2 r47h、abca7 g1527a显示出比ahn系略有增加,而apoe e2/e4衍生的小胶质细胞与ahn衍生的小胶质细胞相比增加了7倍。
[0291]
apoe e4/e4、ap0e e2/e4、trem2 r47h、abca7 g1527a和cd33(带有rs429358 snp)
衍生的小胶质细胞释放略高水平的可溶性分形趋化因子,一种通过降低神经炎症期间的tnf-α和一氧化氮水平促进趋化性、存活和增强神经保护作用的可溶性趋化因子。
[0292]
apoe e4/e4、ap0e e2/e4和abca7 g1527a衍生的小胶质细胞响应lps和dbu-camp释放高水平的cxcl1/groα,而trem2r47h和cd33衍生的小胶质细胞显示释放的这种细胞因子的水平的略微增加,意味着这种分析物与apoe和abca7基因型之间存在相关性。最近的一项研究表明,cxcl1可能促成ad发展中的炎症反应,但不能作为在该疾病的发病机制中赋予ad易感性的潜在遗传因素。在生理条件下,cx3cr1通过限制小胶质细胞的激活来维持小胶质细胞稳态。刺激后释放的这种细胞因子的高水平表明通过cx3cr1的拯救机制的发作以保持与apoe或abca7 g1527a基因型相关的疾病相关小胶质细胞中的稳态功能。或者,apoe e4/e4和abca7 g1527a激活的小胶质细胞分泌的高水平cx3cr1可能是促进神经元变性的信号(atagi等人,2015;wolfe等人,2018)。
[0293]
apoe e4/e4和ap0e e2/e4衍生的小胶质细胞响应lps和il4/dbu-camp释放高水平的il-6,而所有其他基因型分泌与ahn相似的il-6水平。il-6分泌吸引粒细胞,促进细胞介导的体液th2反应并引发炎症。这种机制将再次支持与apoe e4/e4基因型相关的更大的神经炎症。
[0294]
abca7 g1527a衍生的小胶质细胞响应lps释放高水平的il-1β和il-1α,而其他基因型分泌与ahn小胶质细胞相当的水平。
[0295]
与dbu-camp相比,apoe e4/e4、ap0e e2/e4和abca7 g1527a衍生的小胶质细胞也响应lps释放高水平的il-8/cxcl8。abca7 g1527a衍生的小胶质细胞释放高水平的il-8,这意味着促成脑损伤的促炎反应的开始。
[0296]
小胶质细胞还分泌蛋白水解酶和基质金属蛋白酶,其可以消除aβ沉积并限制ad过程,从而在ad中发挥神经保护作用。与ahn衍生的小胶质细胞相比,apoe e4/e4、ap0e e2/e4、trem2 r47h、abca7 g1527a和cd33(带有rs429358snp)衍生的小胶质细胞释放略有升高的mmp-9和mmp-12水平。
[0297]
当受到lps或m1刺激的刺激时,r47h trem2衍生的小胶质细胞与ahn衍生的小胶质细胞相比释放超过7倍的il-12p70。另一方面,响应il4 dbu-camp或m2刺激,ap0e e2/e4与ahn衍生的小胶质细胞相比释放超过7倍的il-12p70。其他基因型显示il-12分泌水平的略微增加。初级小胶质细胞在大脑中产生il-12,以控制感染期间或cns的th1细胞介导的自身免疫性疾病中的免疫反应。r47h trem2衍生的小胶质细胞的增加的il-12水平意味着nk细胞和t细胞的细胞毒性活性的强烈激活。
[0298]
最后,apoe e4/e4、ap0e e2/e4、abca7 g1527a衍生的小胶质细胞释放相似或略高水平的il-13、il-18、il-23和α突触核蛋白水平,暗示缺乏这些分析物与上述基因型的相关性。
[0299]
神经炎症是阿尔茨海默病(ad)发病机制和进展的重要促成因素。几种炎症介质的组合产生与特定snp突变相关的独特特征。来自疾病特异性供体的ipsc衍生的小胶质细胞可用于确定与各种小胶质细胞snp和突变相关基因型相关的关键特征细胞因子。
[0300]
小胶质细胞的吞噬功能对于保持神经保护作用很重要。小胶质细胞介导的吞噬作用可能被疾病特异性snp或突变损坏,这进而可影响大脑中的关键稳态机制。在存在phrodo标记的细菌金黄色葡萄球菌和淀粉样蛋白β的情况下评估疾病相关小胶质细胞(dam)的吞
噬功能,以比较疾病相关snp对小胶质细胞的吞噬功能的作用。此功能可用于高通量筛选应用。
[0301]
在这些小胶质细胞表达的疾病相关基因中,编码在髓样细胞上表达的触发受体2(trem2)和apo e同种型的基因中的序列变体与ad的异常增加的风险有关。apoe是大脑中的主要胆固醇载体,并且在脂质运输、胆固醇稳态和突触稳定性中起着至关重要的作用。抗apoe免疫疗法抑制淀粉样蛋白的积累和沉积,进一步支持apoe在aβ聚集和清除中的作用。已显示apoe表达在疾病相关小胶质细胞中显著上调。已显示apoe4/e4同种型通过在体外上调运动和吞噬行为来固有影响小胶质细胞的生理学。已显示与其他同种型相比,apoe4/e4过表达aβ的减少的摄入。已显示apoe2(第三种最常见的主要apoe同种型)在神经变性疾病中的作用为延迟家族性ad中的疾病发作。迄今为止,尚未彻底研究对ipsc衍生的小胶质细胞功能的apoe同种型特异性作用。
[0302]
trem2感知脂质并介导髓鞘吞噬作用。trem2的功能失去(lof)变体与增加的淀粉样蛋白斑块播种、减少的淀粉样蛋白聚集相关并且与apoe的增加的相互作用触发信号传导级联,导致淀粉样蛋白斑块中减少的小胶质细胞聚集和apoe积累并伴有功能障碍。
[0303]
在细菌金黄色葡萄球菌和淀粉样蛋白β的存在下评价疾病相关小胶质细胞(dam)的吞噬功能,以比较疾病相关snp对小胶质细胞吞噬功能的作用。
[0304]
以同样的方式,在全基因组关联研究中,atp结合盒转运蛋白a7(abca7)已被鉴定为晚发性阿尔茨海默病的易感因素。已显示abca7介导吞噬作用并影响膜运输。abca7与ad密切相关。淀粉样蛋白-β的吞噬清除在abca7-/-小鼠中受损。来源于患者的ipsc具有与abca7中的g1527a取代相关的错义变异,提供bca7在调节大脑中的aβ稳态以改变吞噬细胞功能中发挥作用。
[0305]
cd33是通过调节小胶质细胞中的吞噬作用与阿尔茨海默病(ad)易感性相关的免疫调节受体。trem2在调节小胶质细胞生理和代谢中在cd33下游相互作用,因此表达wt和cd33 rs3865444 snp的ipsc衍生的小胶质细胞可用于验证cd33与受损的吞噬功能相关的作用(caldeira等人,2017)。
[0306]
将冷冻保存的ahn和dam小胶质细胞解冻,在成熟培养基中培养三天,并暴露于phrodo标记的淀粉样蛋白β和phrodo金黄色葡萄球菌。吞噬作用的动力学使用incucyte活细胞分析系统进行测量。总红色物体综合强度用于定量功能反应。
[0307]
与ahn小胶质细胞相比,trem2 r47h小胶质细胞和abca7-g1527a小胶质细胞显示出对细菌金黄色葡萄球菌的强吞噬能力。cd33(带有rs429358snp)小胶质细胞显示与ahn小胶质细胞相比相当的对细菌金黄色葡萄球菌的吞噬能力。trem2 r47h小胶质细胞、abca7-g1527a小胶质细胞和cd33(带有rs429358 snp)小胶质细胞显示与ahn小胶质细胞相比在存在淀粉样蛋白β的情况下降低的吞噬作用强度。
[0308]
与ahn小胶质细胞相比,cw13030ee1 apoe 4/4显示出对金黄色葡萄球菌和淀粉样蛋白β的更高强度的吞噬作用。与ahn小胶质细胞相比,cw13098aa1 apoe 4/4显示出对金黄色葡萄球菌和淀粉样蛋白β的较低强度的吞噬作用。与ahn衍生的小胶质细胞相比,cw13005aa1 apoe 2/4小胶质细胞显示出对淀粉样蛋白β的强吞噬能力和对金黄色葡萄球菌的降低的吞噬作用。cw13074aa1 apoe 4/4小胶质细胞显示出与ahn小胶质细胞相似趋势的对金黄色葡萄球菌的吞噬作用,和对ahn小胶质细胞系的对淀粉样蛋白β的略有增强的吞
噬作用。
[0309]
靶向稳态、促炎和抗炎小胶质细胞亚型的遗传改变的全面理解可以提供新的生物学见解并促进用于神经变性疾病的免疫调节治疗方法的靶标优先排序。
[0310]
实施例9
–
小胶质细胞的额外表征
[0311]
已知细胞外核苷酸例如atp和adp引发受体介导的途径,称为“嘌呤能信号传导”途径。诸如组织稳态、伤口愈合、神经变性、免疫、炎症和癌症的生理过程受嘌呤能信号传导的调节。细胞外atp和p2受体对小胶质细胞激活机制很重要。p2x受体是与atp或其衍生物结合的离子营养型受体。p2y受体之一是响应adp的g蛋白偶联受体。在病理条件下,核苷酸例如atp从受损细胞中释放或泄漏,并用作“找到我”或“吃我”的信号以引起过程延伸、趋化和通过小胶质细胞的吞噬作用。p2受体激活还诱导从小胶质细胞产生细胞因子,包括白介素1b(il-1b)、白介素6(il-6)和肿瘤坏死因子α(tnfα)。这种促炎介质已被显示动态改变星形胶质细胞中的g蛋白偶联受体(gpcr)表达和功能。
[0312]
表征了小胶质细胞的嘌呤能受体反应。小胶质细胞在小胶质细胞分化培养基中解冻并复原为200,000个细胞/孔的细胞悬液。在384孔板的每孔中加入15μl小胶质细胞悬浮液。用不同剂量(0-1000nm)的bzatp和adp处理细胞。还在az11645373(p2x7拮抗剂)和azd1283(p2y
12
受体的强效拮抗剂)的存在下测量了对bzatp和adp的反应。对于所有处理,向细胞加入化合物或抑制剂的10ul的4x储备液。在测定前,细胞在抑制剂的存在或不存在的情况下暴露于化合物30分钟。将一瓶flpr calcium-6用测定缓冲液b复原至11ml,并将15μl染料溶液在存在化合物的情况下添加到细胞悬液中。细胞在37℃下孵育1.5小时,并在fdssμcell系统上进行成像。
[0313]
图44a显示了具有atp/bzatp所有迹线的小胶质细胞,图44b显示了具有atp/bzatp样品迹线的小胶质细胞。图44c-f显示了在p2x7拮抗剂az11645373的存在下小胶质细胞对bzatp、adp、bzatp的反应和在p2x7拮抗剂a438079的存在下对bzatp的反应。图44g显示了剂量依赖性反应以证明在azd1283的存在下小胶质细胞中的功能性adp依赖性反应。
[0314]
实施例10
–
从ipsc产生神经前体细胞
[0315]
体外疾病模型的成功开发取决于源自患者衍生的ipsc的大量末期谱系的可用性。神经前体细胞(npc)是自我更新的祖细胞,具有生成神经元和神经胶质的能力(breunig等人,2011)。用于从原代神经细胞和ipsc生成npc有许多效率各不相同的已建立的方案(shi等人,2012a,shi等人,2012b)。大多数最近的方案依赖于smad信号传导途径的抑制。本技术描述了一种简单的方案以利用ipsc向外胚层的自发漂变而不使用双smad抑制途径在不同的ipsc系中生成npc。产生这种细胞类型的基本原理是将其与ipsc衍生的小胶质细胞配对以产生长期共培养测定,以在来源于正常和疾病特异性ipsc细胞的盘中模拟人脑发育和神经谱系、小胶质细胞、内皮细胞、周细胞和星形胶质细胞之间复杂的细胞间相互作用。
[0316]
在该研究中,在matrigel
tm
、层粘连蛋白或玻连蛋白包被的平板和e8培养基上维持多个附加型重编程的ipsc系。在分化开始之前,将ipsc维持在缺氧条件下,以产生神经前体细胞。为了启动神经前体分化,收获ipsc并在rock抑制剂存在的情况下使用e8培养基以15k/cm2铺板到matrigel
tm
、层粘连蛋白或玻连蛋白平板上。将细胞在不存在rock抑制剂的情况下置于新鲜的e8培养基中持续接下来的48小时。下一步涉及预处理步骤,包括将ipsc培养物在常氧条件下置于补充有3um chir的dmemf12培养基中72小时,每天更换培养基。在
预处理步骤结束时收获细胞,然后以30k/cm2以2d格式重新铺板在matrigel
tm
、层粘连蛋白或玻连蛋白板上,或使用超低附着板或旋转瓶在rock抑制剂的存在下以每毫升30万个细胞的密度生成3d聚集体。在接下来的8天,培养物在常氧条件下每隔一天用补充有n2的e6培养基进行补料。在分化的第14天,收获培养物并使用tryple进行个体化。通过用于流式细胞术的细胞表面染色针对tra-162、cd56、cd15的存在和通过用于流式细胞术的细胞内染色针对sox1、神经上皮干细胞蛋白、β3微球蛋白和pax-6表达的存在对细胞进行染色。涉及生成npc的不同步骤在图45a中概述。在三个ipsc系中在不同分化天数出现的npc标志物总结在图45b中。cd56被用作通过这种方法获得的npc的标志物。使用cs10冷冻保存细胞,并且它们在解冻后保持纯度和增殖潜能。npc被置于下游分化方案中以生成星形胶质细胞和pan神经元。
[0317]
根据julia等人概述的方案,从npc分化星形胶质细胞。第14天的npc细胞以15k/cm2在科学细胞星形胶质细胞培养基(science cell astrocyte medium)中铺板到matrigel
tm
包被的6孔板上。每两天对平板进行完整的培养基交换。每6天,或当培养物为约90%汇合时,使用accumax收获平板并以15k/cm2重新铺板到matrigel
tm
包被的6孔板上。如上所述对培养物进行补料并重新铺板持续4代。在4代结束时,针对表面标志物cd44和谷氨酸天冬氨酸转运蛋白(glast)和细胞内标志物胶质纤维酸性蛋白(gfap)、兴奋性氨基酸转运蛋白1(eaat1)、谷氨酰胺合成酶(gs)、水通道蛋白4(aqp4)和s100钙结合蛋白b(s100β)对培养物进行染色(图45c)。
[0318]
皮质谷氨酸能神经元使用由slosarek等人开发的方案从npc产生。第14天npc被铺板在补充有1μm环amp、10ng/ml脑源性神经营养因子(bdnf)和10ng/ml胶质源性神经营养因子(gdnf)的e6培养基中30天。随后将培养基改为皮质神经分化培养基(e6培养基、1μm环amp、10ng/ml bdnf、10ng/ml gdnf、100ng/ml胰岛素样生长因子-i和2%b27补充剂)持续额外的30天(brennan等人,2011)。对于不同的ipsc系,在第14-36天之间观察到皮质谷氨酸能神经元。通过针对β3微管蛋白、map2表达的存在进行染色来确认神经培养物的纯度。
[0319]
***
[0320]
根据本公开内容,无需过度实验即可做出和进行本文公开和要求保护的所有方法。虽然本发明的组合物和方法已经根据优选实施方案进行了描述,但对本领域技术人员来说明显的是,可以对本文所述方法和本文所述方法的步骤或步骤顺序应用变化而不脱离本发明的概念、精神和范围。更具体地,很明显,某些化学和生理学相关的药剂可以替代本文所述的药剂,同时将获得相同或相似的结果。所有这些对本领域技术人员来说明显的类似替代和修改都被认为在由所附权利要求书限定的本发明的精神、范围和概念之内。
[0321]
参考资料
[0322]
以下参考资料,在它们提供示例性程序或补充在本文阐述的那些的其他细节的程度上,通过引用具体并入本文。
[0323]
atagi等人,j biol chem.290(43):26043-50,2015.
[0324]
brennand等人,2011
[0325]
caldeira等人,front aging neurosci.9:277,2017.
[0326]
cheng等人,clin chim acta.463:88-95,2016.
[0327]
国际专利公开号wo 02/016536
[0328]
国际专利公开号wo 03/016496
[0329]
国际专利公开号wo 98/30679
[0330]
国际专利公开号wo 98/53058
[0331]
国际专利公开号wo 98/53059
[0332]
国际专利公开号wo 98/53060
[0333]
julia等人,stem cell reports.9(2):600-614,2017.
[0334]
mcquade等人,j mol biol.431(9):1805-17,2019.
[0335]
melief等人,glia.60(10):1506-17,2012.
[0336]
minett等人,j neuroinflammation.13(1):135,2016.
[0337]
pct公开号2012/149484
[0338]
rustenhoven等人,trends in pharmacological sciences,38(3),291-304,2017.
[0339]
slosarek等人,cell rep.24(9):2248-2260,2018.
[0340]
美国专利申请号12/715,136
[0341]
美国专利号6,140,081
[0342]
美国专利号6,453,242
[0343]
美国专利号6,534,261
[0344]
美国专利号6,617,152
[0345]
美国专利号8,372,642
[0346]
美国专利公开号2002/0076747
[0347]
美国专利公开号2005/0064474
[0348]
美国专利公开号2005/0260186
[0349]
美国专利公开号2006/0104968
[0350]
美国专利公开号2006/0188987
[0351]
美国专利公开号2007/0218528
[0352]
美国专利公开号2011/0301073
[0353]
美国专利公开号2011/0301073
[0354]
美国专利公开号2011/0301073.
[0355]
westin等人,plos one.7:e30525,2012.
[0356]
winkler等人,brain pathology(zurich,switzerland),24(4),371
–
386,2014.
[0357]
wolfe等人,int j mol sci.20(1),2018.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。