特异性检测hiv-2前病毒dna的引物及其应用
技术领域
1.本发明属于分子生物检测领域,更具体涉及对hiv-2前病毒dna的检测。
背景技术:
2.人类获得性免疫缺陷综合征(aids)又称为艾滋病,是由感染艾滋病病毒(hiv)所引起的一种危害性极大的传染病。hiv是一种攻击人体免疫系统的逆转录rna病毒,通过破坏cd4 t淋巴细胞,使人体失去免疫功能,从而易于感染各种疾病并导致死亡(1)。艾滋病起源于非洲,通过移民带入美国,在1981年由美国首次报导,后迅速蔓延至各国。根据基因型差异,hiv可以分为hiv-1和hiv-2两个大型,多种亚型。目前世界上大部分地区流行的是hiv-1,而hiv-2只在西非、欧洲(尤其是法国和葡萄牙)等地区流行(2,3)。
3.与hiv-1的高传染性、高致死性相比,hiv-2被认为更像一种温和的变体。有研究报导,在没有接受抗逆转录病毒治疗前提下,98%以上的hiv-1感染者会患上艾滋病;而大部分hiv-2患者都会有正常的预期寿命,而且没有相关并发症,因此人们长期以来普遍认为hiv-2不会像hiv-1一样导致疾病(4)。而且由于hiv潜伏期较长、发病地区较窄,使得人们对开发诊断和治疗hiv-2的兴趣不强烈。但对1990年至2013年4900名患者进行长期随访(包括血液测试)的报导指出,hiv-2患者遭受hiv相关感染和患上艾滋病的方式与hiv-1几乎是相同的,只是其疾病进展相对缓慢(5)。最近研究表明,即使疾病进展较缓,hiv-2患者的预期寿命比正常未感染者少10年,且43%的hiv-2感染者会患上艾滋病(6)。因此,hiv-2比之前认为的更具致命性,开发诊断性试剂和治疗性药物需引起人们重视。
4.hiv-2基因组由两条单链正链rna组成,包含gag、pol、env三个结构基因以及tat、rev、nef、vif、vpr、vpx等调控基因,在基因组的5’和3’端各有一段长末端重复序列(ltr),富含顺式调控元件并控制病毒基因的表达。各基因功能不同,其中env基因编码的863个氨基酸的前体蛋白可以糖基化为gp160、gp120和gp41,尤其是gp120含有中和抗原决定簇,在病毒与细胞融合中起重要作用;另外,env基因变异率最高,根据其基因区的突变又可将hiv-2分为8个亚型(a-h)(7)。亚型a(发病人群主要为大西洋临近国家如几内亚)和亚型b(发病人群主要为北宁湾临近国家如加纳)的hiv-2与临床相关,其余亚型只发生在少部分地区(8)。
5.自hiv-2发现30多年以来,其并未广泛传播,而且基本上为输入性病例(如药物注射或者具有性接触史),在2018年湖南省首次诊断出2例非输入性hiv-2感染病例(9)。hiv-2传播地区较窄的主要原因是其较低的病毒载量(lower viral load)和复制速率(lower viral replication rate),这会导致其在血浆中的rna病毒较低甚至检测不到,同时会导致其较低的传播性及较长的无症状期(10,11),对病毒的发现和诊断带来了很大的阻碍。而且,虽然hiv-2的致病力低于hiv-1,但一些可有效抑制hiv-1的药物如非核苷酸反转录酶抑制剂和蛋白酶抑制剂对hiv-2治疗无效(12),因此区分hiv-1和hiv-2对于临床治疗十分重要。
6.体外检测hiv感染的最常用方法为gag抗原捕捉elisa实验(13),适用于细胞培养
上清或细胞裂解物中gag抗原的检测,实验中不依赖特异病毒或细胞系的使用,实验方法简单快速,但此方法最大的缺陷就是需要获得相匹配的抗体去量化病毒蛋白。除此之外,常用的hiv检测方法还有免疫印迹实验(wb)和pcr检测。目前我国检测hiv抗体的常规程序是初筛-复筛-确认,常用的确认试验方法为wb。根据2004年《全国艾滋病检测技术规范》说明,对在hiv-1/2混合型wb检测中出现hiv-2特异性指示带的样品,需用hiv-2型免疫印迹实验再做hiv-2抗体确认实验,呈阳性反应后再将样品送国家参比实验室进行核酸序列分析。然而,由于wb检测会产生大量的不确定甚至假阳性结果(14),以及我国目前对hiv-2抗体确认试剂盒的评价工作滞后,缺乏标准化的hiv-2核酸检测方法,hiv-2的诊断工作未能广泛开展。
7.荧光定量pcr技术广泛应用于动物疾病检测、肿瘤基因检测、遗传基因检测和分子生物学定量研究等多个领域。荧光定量pcr技术是在pcr反应体系中加入荧光基团,随pcr产物积累,荧光信号不断增强并积累成一条扩增曲线,通过荧光积累曲线实时监测整个pcr进程并对未知模板进行定性和定量分析的方法。qrt-pcr(quantitative real-time pcr)技术主要包括扩增序列非特异性检测和扩增序列特异性检测两大类。扩增序列非特异性检测主要基于与dna结合的荧光分子如sybr green i染料,染料渗入dna小沟并能发出荧光信号。但由于其不能与单链dna或rna结合,以及其可以与任何双链dna结合而产生非特异性信号,使得检测的准确性和特异性受到限制。扩增序列特异性检测又分为直接法和间接法,其中直接法是指标记荧光的探针与扩增产物结合后直接产生荧光,如分子信标、荧光标记引物和分子探针,但这种方法使用较少。间接法即利用水解探针策略(如taqman系统),在探针的5’端添加荧光报告基团(如fam等)并在3’端添加淬灭基团。由于完整的探针受到激发光后会发生荧光共振能量转移,因此检测不到信号;但在pcr扩增过程中,引物/探针会与模板结合,在taq酶的5
’-3’
外切酶活性下将探针5’端的荧光基团切割下来,从而脱离淬灭基团的屏蔽并进而能接收到荧光信号。由于被释放的荧光基团数目和pcr产物数量是一对一的关系,因此荧光信号的强弱代表了模板的数量,从而实现了对模板的定量(15)。
8.因此,由于传统血清学检测的弊端:如不能在感染窗口期检测出hiv-2,以及hiv-2与hiv-1在血清学上存在的交叉反应,灵敏度高、特异性强的qpcr技术为hiv-2的检测带来了诸多便利。研究人员已经开始使用qrt-pcr技术检测血浆中的hiv-2rna病毒,可以检测至500copy/ml血浆(16)。但据法国国家艾滋病研究署报导,36%的艾滋病病人检测不出血浆中的hiv-2rna(<100copy/ml)(17),这主要是由于病人的发病级数不同导致血浆中rna病毒的含量不同。即使随着pcr技术的发展,如仪器的更新、高效的pcr酶等试剂,人们可以检测出血浆中相对低含量的hiv-2rna病毒(18),但仍有病人样本中的rna检测不出。
9.前病毒(provirus)是由病毒的rna逆转录为dna,整合在宿主染色体内所形成的一部分。以hiv-1的rna和前病毒dna为模板进行扩增实验发现,以前病毒dna为模板能显著提高扩增阳性率(19)。而且dna样本比rna更加稳定,可以在低温条件下长期储存;来源广泛(可以是各种组织器官或血液,而不局限于血浆)、提取和处理也相对简单,因此检测体内的hiv-2前病毒dna是进行诊断的更好方法(20)。目前,对hiv-2前病毒dna量化的主要限制为缺乏dna标准。
技术实现要素:
10.本发明以hiv-2保守的ltr区序列为靶标,设计合成了高效特异的hiv-2引物/探针,并以其扩增序列构建质粒作为dna标准,可以精确并特异性地检测出hiv-2的前病毒dna,而与其它来源(如人、猴、兔、大鼠、小鼠)的dna不会有任何交叉反应。
11.本发明的目的是提供hiv-2前病毒dna特异性的引物和/或探针,以及可用于检测样本中hiv-2前病毒dna的taqman qpcr方法,此方法适用于检测样本中是否存在hiv-2前病毒dna。
12.术语“hiv-2”是引起获得性免疫综合征的病毒(又称艾滋病病毒)2型,是一种逆转录rna病毒。该病毒的rna感染宿主后经逆转录合成相应的dna并整合进入宿主基因组dna。
13.术语“hiv-2前病毒dna”是指hiv-2病毒感染宿主细胞后,逆转录合成并整合到宿主基因组上的dna。本发明中,“hiv-2基因片段”或“hiv-2片段”可以互换使用,用于指hiv-2前病毒dna的片段。
14.本发明一方面提供特异性检测hiv-2前病毒dna的引物对,所述引物对包括正向引物5'-caggtagagcctgggtgttc-3'(seq id no:1)和反向引物5'-ggtctttaagcaagcaagcgt-3'(seq id no:2)。
15.本发明的另一方面提供特异性检测hiv-2前病毒dna的引物和探针组合,所述引物包括正向引物和反向引物,其中正向引物是5'-caggtagagcctgggtgttc-3'(seq id no:1),所述反向引物是5'-ggtctttaagcaagcaagcgt-3'(seq id no:2);所述探针的序列为5'-ttggccggtactgggcagacggctcc-3'(seq id no:3),该探针的5’端标记报告荧光基团,3’端标记淬灭基团。
16.在一些实施方案中,所述荧光探针的5’端标记的报告荧光基团为fam,3’端标记的淬灭基团为nfq-mgb或tamra。
17.本发明的另一方面提供上述引物对或引物和探针组合在检测样本中hiv-2前病毒dna中的应用。
18.本发明的另一方面提供上述引物对或引物和探针组合在制备用于检测样本中hiv-2前病毒dna的试剂中的应用。
19.在一些实施方案中,所述检测是通过qpcr检测或通过数字pcr检测。
20.本发明中所述的qpcr是指实时荧光定量pcr(real-time quantitative pcr),是在pcr反应体系中加入荧光基团,利用荧光信号积累实时监测pcr进程,最后可以通过标准曲线对未知模板进行定量分析。在qpcr检测中,ct值表示循环阈值,即每个反应管内的荧光信号达到设定阈值时所经历的循环数。由于每个模板的ct值与该模板的起始含量的对数存在线性关系,起始拷贝数越多,ct值越小。利用连续稀释的已知起始含量的标准品可作出标准曲线,其中横坐标代表起始含量的对数,纵坐标代表ct值,或者纵坐标代表起始含量的对数,横坐标代表ct值。只要获得未知样品的ct值,即可从标准曲线上计算出该样品的含量。qpcr在本领域中属于成熟技术,利用现有的仪器进行qpcr检测时,可以直接从仪器的输出结果中获得样本的ct值。
21.在qpcr检测中,可以使用荧光探针或荧光染料获取荧光信号。常见的荧光探针例如可以是taqman荧光探针,其中pcr扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。探针完
整时,报告基团发射的荧光信号被淬灭基团吸收;pcr扩增时,taq酶的5'-3'外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条dna链,就有一个荧光分子形成,实现了荧光信号的累积与pcr产物形成完全同步。在一些实施方案中,报告荧光基团可以是例如fam,淬灭基团可以是例如nfq-mgb或tamra。本领域技术人员知道其它报告荧光基因和相应的淬灭荧光基团也可以用于本发明。
22.在qpcr检测中,还可以使用荧光染料获取荧光信号,例如可以在pcr反应体系中,加入过量荧光染料,荧光染料非特异性地掺入dna双链后,发射荧光信号,而不掺入链中的染料分子不会发射任何荧光信号,从而保证荧光信号的增加与pcr产物的增加完全同步。常用的荧光染料例如可以是sybr荧光染料、磺酰罗丹明(texas red)、异硫氰酸荧光素(fitc)、羟基荧光素(fam)、四氯荧光素(tet)、joe、vic、rox和ned等。
23.数字pcr检测也是本领域技术人员公知的,简言之,数字pcr(也可称单分子pcr)包括pcr扩增和荧光信号分析,在pcr扩增阶段,将样品稀释到单分子水平并平均分配到几十至几万个单元中进行反应,在扩增结束后对每个反应单元的荧光信号进行采集。最后通过直接计数或泊松分布公式计算得到样品的原始浓度或含量。本领域技术人员熟知如何进行数字pcr检测。
24.本发明的另一方面提供特异性检测样本中hiv-2前病毒dna的试剂盒,该试剂盒包括上述引物对,或包括上述引物和探针组合。
25.在一些实施方案中,所述试剂盒中还包括进行qpcr检测所需要的任意一种或更多种试剂。
26.在一些实施方案中,所述进行qpcr检测所需要的任意一种或更多种试剂包括选自下述组分的一种或更多中组分:qpcr反应液(例如qpcr master mix(2
×
),其中包括qpcr反应所需要的酶等必须成分)、水(例如无核酸酶高纯水)、对照品。其中对照品可以是阳性对照品,例如hiv-2前病毒标准品,如含hiv-2前病毒dna的质粒dna,和/或阴性对照品。其中阳性对照品还可以是具有hiv-2前病毒dna的质粒dna、基因组dna或细胞。其中阴性对照品可以是无hiv-2前病毒dna的质粒dna、基因组dna、细胞或水。所述试剂盒中还可以包含hiv-2前病毒dna标准品,例如具有hiv-2前病毒dna片段的质粒dna。
27.本发明的另一方面提供检测样本中hiv-2前病毒dna的方法,包括使用上述的引物和探针组合,对从样本提取的dna进行qpcr检测,根据qpcr检测结果定性检测或定量检测样本中是否存在hiv-2前病毒dna。从样本提取的dna可以例如是基因组dna。
28.本发明的另一方面提供检测样本中hiv-2前病毒dna的方法,包括以下步骤:
29.(1)提取样本中的dna;
30.(2)使用特异性扩增hiv-2前病毒dna的引物和探针组合,例如上述的引物和探针组合,对hiv-2前病毒dna标准品和样本dna进行qpcr检测,其中hiv-2前病毒dna标准品是用含有hiv-2前病毒dna片段的dna配制的不同给定浓度的样品;
31.(3)用hiv-2前病毒dna标准品的qpcr检测结果制作标准曲线,拟合线性方程,其中r2≥0.99;且各标准品浓度的准确度(re%)为-75%~150%,确定可稳定检测到的浓度最低点;
32.(4)结果判定:如果样本dna的qpcr结果中出现明显的扩增曲线;且样本dna的qpcr的ct值小于浓度最低点的ct值,则为阳性结果,即样本中存在hiv-2前病毒dna;如果无明显
扩增曲线,或者有明显扩增曲线,但ct值大于标准曲线浓度最低点的ct值,则为阴性结果,即样本中不存在hiv-2前病毒dna。
33.在一些实施方案中,所提取的是样本中的基因组dna。所述样本dna可以是样本基因组dna。从样本中提取基因组dna的方法是本领域公知的,例如可以用dna提取试剂盒提取基因组dna。
34.在一些实施方案中,上述步骤(3)中制作标准曲线时,以hiv-2前病毒dna标准品的ct值为纵坐标(y),以hiv-2前病毒dna标准品的dna浓度的对数为横坐标(x),拟合线性方程。
35.在一些实施方案中,上述hiv-2前病毒dna标准品包含至少6个用含有hiv-2前病毒dna片段的dna配制的不同给定浓度的样品。hiv-2前病毒dna标准品的数量可以是6个、7个、8个或更多个。hiv-2前病毒dna标准品例如可以用含hiv-2前病毒dna片段的质粒dna与水混合配制而成。
36.拟合标准曲线时,如r2和各标准品浓度的准确度不满足以上要求,可以通过重复实验,或重新制备dna标准品、或换用不同浓度的dna标准品,来获得满意的拟合结果。标准曲线的拟合以及获得满意拟合结果的方法属于本领域技术人员的公知技术。
37.浓度最高点是指用上述方法可稳定检测到的浓度的最高限,在本发明中也可称为检测上限或定量上限。可稳定检测到的浓度的最低限即为浓度最低点,在本发明中也可称为检测下限或定量下限。
38.判断是否出现明显扩增曲线的方法是本领域技术人员公知的,例如当δrn vs cycle模式下的曲线为s形时,可确定出现明显扩增曲线。
39.在一些实施方案中,qpcr的反应体系为pcr master mix(2
×
)10μl,特异性扩增hiv-2前病毒dna的引物/探针(20
×
)1μl,dna样品 无核酸酶高纯水9μl。其中dna样品可以是样本dna、dna标准品、或其它对照品、阴性对照品或质控品。
40.在一些实施方案中,qpcr反应的程序为首先50℃,2min以激活udg;然后95℃,10min激活dna聚合酶;然后按下列参数进行40个循环:95℃,15秒;60℃,1min。在一些实施方案中,在applied biosystems abi 7500real time pcr仪上完成qpcr反应。
41.在一些实施方案中,所述结果判定也可以是定量检测样品中的hiv-2前病毒dna,或者可以进一步包括定量检测样品中的hiv-2前病毒dna。所述定量检测可以包括例如根据样本dna的ct值和拟合的标准曲线,确定样本dna中hiv-2前病毒dna的浓度。
42.本发明中,样本例如可以是器官、组织、全血、细胞、体液样品或生物制品。
43.本发明中,样本例如可以来源于人或任何动物,例如小鼠、大鼠、兔、猴等动物,或者任何其他途径。
44.在一些实施方案中,本发明的方法是在体外进行的。
45.在一些实施方案中,本发明的方法是非诊断性的,例如可以用于确定生物制品,例如疫苗、抗体或细胞制品是否受到hiv-2污染。
46.本技术中,术语“hiv-2阳性的”、“含有hiv-2前病毒dna的”,“受hiv-2感染的”或者类似的表述,是指受到hiv-2感染的,基因组dna中含有hiv-2前病毒dna的细胞或含有这些细胞的组织、器官。
47.本发明提供了新的hiv-2检测手段,应用实时荧光定量pcr(qpcr)技术检测整合于
宿主基因组中的hiv-2前病毒dna片段,并提供针对该dna片段的特异引物和/或探针。本发明的引物、探针和方法能够大大提高检测样本中hiv-2的灵敏性和特异性,可用于追踪并定性或定量检测样本中hiv-2前病毒dna,用于检测样本是否感染人hiv-2病毒,或者区分hiv-1患者是否具有hiv-2双重感染,并能够用于动物模型中hiv-2核酸药物或疫苗研究过程中动物器官、组织、细胞和血液中hiv-2前病毒dna的追踪和定量检测。
附图说明
48.图1是标准品质粒的酶切图谱。
49.图2是标准品质粒的酶切验证结果。
50.图3是hiv-2引物/探针灵敏性验证扩增曲线图。
51.图4是hiv-2引物/探针标准曲线线性图。
52.图5是hiv-2引物/探针特异性和普遍适用性验证扩增曲线图。
53.图6是hiv-2引物/探针线性验证扩增曲线图。
实施例
54.下面通过实施例,并结合附图,对本发明的技术方案作进一步详细的说明,但本发明不限于下面的实施例。
55.如无特别说明,下述实施例中的qpcr实验方法均为taqman qpcr方法,提及的序列均以从5’端到3’端的方式表示。
56.1.材料与方法
57.1.1引物/探针的设计
58.根据ncbi基因数据库信息,以hiv-2前病毒相对保守的5’端长末端重复序列(ltr)作为模板设计并合成引物及荧光探针(由thermo fisher scientific,waltham,ma,usa合成),设计好的引物/荧光探针通过blast对比分析,选择其中只扩增hiv-2病毒而与其它病毒无交叉反应的引物序列。引物和探针信息如下表所示:
59.引物名称hiv-2gene id9628880正向引物caggtagagcctgggtgttc反向引物ggtctttaagcaagcaagcgt探针fam-ttggccggtactgggcagacggctcc-nfq-mgb
60.1.2hiv-2标准品质粒dna的构建
61.将含有扩增序列在内的共200bp的hiv-2前病毒dna片段,使用xbai和bamhi双酶切并克隆至puc57载体,质粒构建成功后经酶切鉴定并去内毒素纯化,获得的纯化质粒dna经nanodrop测定浓度后稀释成标准品使用,其所含有的拷贝数为3.13
×
108copy/ng质粒。质粒图谱如图1所示,酶切结果如图2所示。
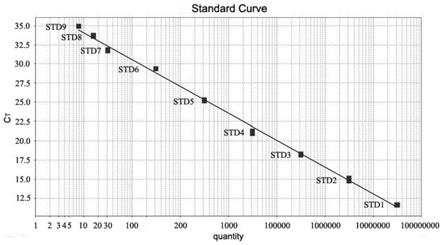
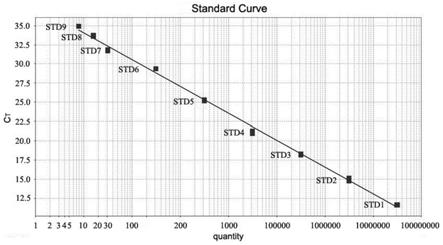
62.1.3样本的选择
63.以上述引物/探针、标准品质粒dna,结合相应的pcr反应所需要的酶系,经taqman qpcr反应可以检测样本dna中的hiv-2前病毒dna。在此检测系统中,我们选择来源于人、食蟹猴、新西兰兔、sd大鼠和小鼠的组织或细胞作为样本,采用qiagen公司的基因组dna提取
试剂盒dneasy blood&tissue kit,按照试剂盒提供的标准程序提取动物组织或细胞基因组dna,进行该qpcr系统的特异性和灵敏性等的检测。
64.1.4荧光定量pcr反应
65.taqman qpcr扩增反应使用abi 7500(applied biosystems)仪器完成,以标准品质粒dna为模板,加入universal pcr master mix(2
×
)、人hiv-2的特异性引物和荧光标记的探针,进行扩增检测。qpcr反应的内在质控和定量由标准品质粒作为标准,使用一定量质粒dna梯度稀释并用于标准曲线验证,通过拷贝数计算和标准曲线的线性标准(r2≥0.99)控制整个qpcr反应的进行。
66.反应体系为:
67.成分体积(μl)pcr master mix(2
×
)10hiv-2引物/探针(20
×
)1样本dna5无rnase/dnase水4
68.pcr反应程序为:首先50℃反应2min激活udg,然后95℃运行10min以彻底激活dna聚合酶,最后95℃,15sec和60℃,1min,经40次循环扩增模板dna,并对样本中的hiv-2前病毒dna进行定量。
69.1.5taqman qpcr反应检测限的验证
70.通过对标准品质粒dna进行梯度稀释,以qpcr反应可以检测到的最低含量作为检测下限。设定检测下限为标准曲线的最低点,以经3次以上重复试验可稳定检测到的dna含量用于qpcr反应中标准曲线的最低点。
71.1.6taqman qpcr反应普遍适用性的验证
72.以上述检测限为qpcr反应标准曲线最低点,制作制质粒dna的标准曲线,进行hiv-2的taqman qpcr反应。以此反应体系,验证hiv-2引物的特异性以及此引物与其它样本来源的dna(如正常人、食蟹猴、新西兰兔、鼠等动物来源)有无交叉反应。
73.1.7数据分析
74.实验中所有pcr测定结果都由abi 7500仪器完成,使用7500software v2.4采集,经7500software v2.4和microsoft excel 2007软件进行数据处理,并由adobe photoshop cs6软件进行图片的绘制。另外,实验中数值变异度使用变异系数(cv)表示:cv%=标准偏差/平均数*100%;准确性(re)使用公式:re%=(实际值-理论值)/理论值*100%计算。其中无扩增的样本视为阴性,高于检测下限并有明显扩增曲线的样本视为阳性。
75.2.实验结果
76.2.1hiv-2特异性引物和标记探针的选择
77.由于hiv-2病毒基因的高度可变性,选择相对保守区的基因设计引物/探针可以保证多种病人来源样本中hiv-2的精确检测。经过对hiv-2各基因序列所设计的引物的保守性和敏感性分析(21),我们选择了高度保守的ltr区设计引物/探针,并对其进行ncbi数据库blast检测,检测结果表明此引物/探针与siv或hiv-1以及其它类型的病毒或其它物种的基因序列无同源性,具有高度的特异性。
78.2.2hiv-2特异性引物/探针的灵敏性
79.为了验证hiv-2taqman qpcr方法的灵敏性,将标准品质粒dna(起始100pg,3.13
×
107copy)以10倍梯度稀释,进行定量下限的检测,其扩增曲线如图3所示。实验结果发现,hiv-2引物/探针可检测低至25夸克(ag)或7.8拷贝(copy)的质粒dna,具有较高的灵敏性;同时qpcr反应的扩增效率大于90%。
80.2.3高灵敏性hiv-2taqman qpcr反应体系的建立
81.以hiv-2特异性引物/探针、universal pcr master mix(2
×
)和hiv-2标准品质粒dna建立taqman qpcr反应体系。体系中标准曲线样本由hiv-2标准品质粒稀释而成,以100pg质粒dna为起始浓度,进行样品的10倍梯度稀释,稀释后样品浓度分别为100pg/5μl、10pg/5μl、1pg/5μl、100fg/5μl、10fg/5μl、1fg/5μl、100ag/5μl、50ag/5μl、25ag/5μl。扩增结束后数值结果以ct值为纵坐标(y),以拷贝数的对数为横坐标(x)拟合标准曲线(标准曲线线性图的横坐标仍以标准品原值标示),如图4所示,拟合后标准曲线线性回归方程为:y=-3.509x 37.64;相关系数r2=0.997。以此标准曲线进行待测样本中hiv-2前病毒dna含量的计算。
82.同时,对此qpcr反应体系进行批间变异度分析,在三次独立实验过程中,通过对ct值和标准品质粒dna拷贝数回归标准曲线计算后,批间样本的变异性低,表明实验方法的稳定可靠。
83.表1三次独立实验中各稀释样本平均ct值和批间变异系数比较
[0084][0085][0086]
2.4hiv-2qpcr反应体系的特异性和普遍适用性
[0087]
为了验证hiv-2qpcr反应体系的特异性和普遍适用性,使用hiv-2引物/探针扩增不同拷贝数的质粒dna(从31300000~7.8copy)以及人、食蟹猴、新西兰兔、sd大鼠和小鼠的基因组dna,扩增曲线图如图5所示,发现:hiv-2引物/探针可以特异并稳定地检测出质粒中hiv-2而与上述人和各种动物的基因组dna无交叉反应。
[0088]
以上结果表明,hiv-2qpcr反应体系具有高度特异性,可稳定检测出样本中的hiv-2前病毒,并与其它物种的基因组不会发生任何交叉反应。
[0089]
2.5hiv-2qpcr反应体系的稀释线性
[0090]
为了验证hiv-2特异性引物/探针及qpcr反应体系的线性,将hiv-2标准品质粒dna(起始3.13
×
108copy)分别进行20倍、400倍、8000倍和160000倍的梯度稀释,进行qpcr检
测,扩增曲线图如图6所示,对数值结果分析比较发现:所有实际测得浓度和样品稀释倍数呈线性关系。
[0091]
然后对4个梯度的稀释样本数值进行计算,2组重复取平均值,结果如表2所示:所测实际数值与理论数值相差较小,具有较高的准确性;同时各稀释梯度样本复孔之间的变异度较低。
[0092]
表2稀释线性实验中各样本准确性和变异度比较
[0093]
稀释样本理论拷贝数实际拷贝数准确性(re%)变异度(cv%)20
×
1565000013867578-11.47.9400
×
782500870311.211.27.28000
×
3912537189.67-4.91.5160000
×
1956.251761.137-10.01.0
[0094]
3.临床样本检测
[0095]
在这项研究中,总共检测了52例经血清学检测确诊的hiv-2阳性样品和而119例经血清学检测核实的hiv-2阴性样品。
[0096]
本方法的测试结果和标准对照方法的测试结果如表3所示,其中的“本方法”是按照前述“1.材料与方法”所述的引物、探针、实验方法和“2.实验结果”所述的标准曲线进行检测的方法,其中的“标准对照方法”是指mikrogen recomline hiv-1&hiv-2igg条带免疫法。
[0097]
表3本方法与标准对照方法检测结果比较
[0098][0099]
根据对照方法的检测结果,将受试者分为阳性组和阴性组,并将本方法的检测结果与对照方法结果进行比较。符合率的结果如下:
[0100]
诊断敏感性=52/(52 0)
×
100%=100%
[0101]
诊断特异性=117/(2 117)
×
100%=98.32%
[0102]
总符合率=(52 117)/(52 0 2 117)
×
100%=98.83%。
[0103]
4.讨论
[0104]
hiv-2感染者的血液中病毒载量相对较少,因此需要高敏感性的方法进行检测。发明人发明了hiv-2特异性的taqman qpcr实验方法,可用于检测患者体内的hiv-2前病毒dna,可以在hiv-2患者诊断以及hiv-2疫苗研究中发挥重要作用。
[0105]
之前有研究表明,无论在感染早期还是已经发展成艾滋病时,hiv-2感染者比hiv-1感染者具有较高的cd4细胞数量。这种cd4细胞数量的不同可能与hiv-2的低复制率有关,主要归咎于hiv-2病毒的特性、较低的免疫超活化、较好的免疫控制或免疫系统对其较强的抑制效应(11,22)。因此在缺乏可靠的hiv-2检测试剂盒前提下,人们常采用监控cd4细胞数
19.
[0128]
16.schutten m,van den hoogen b,van der ende me,gruters ra,osterhaus ad,niesters hg.development of a real-time quantitative rt-pcr for the detection of hiv-2 rna in plasma.j virol methods.2000;88(1):81-7.
[0129]
17.drylewicz j,matheron s,lazaro e,damond f,bonnet f,simon f,et al.comparison of viro-immunological marker changes between hiv-1 and hiv-2-infected patients in france.aids.2008;22(4):457-68.
[0130]
18.delarue s,didier e,damond f,ponscarme d,brengle-pesce k,resche-rigon m,et al.highly sensitive plasma rna quantification by real-time pcr in hiv-2 group a and group b infection.j clin virol.2013;58(2):461-7.
[0131]
19.ferrer p,montecinos l,tello m,tordecilla r,rodriguez c,ferres m,et al.hiv-1 tropism:a comparison between rna and proviral dna in routine clinical samples from chilean patients.virol j.2013;10:318.
[0132]
20.behrendt r,fiebig u,norley s,gurtler l,kurth r,denner j.a neutralization assay for hiv-2based on measurement of provirus integration by duplex real-time pcr.j virol methods.2009;159(1):40-6.21.walther l,grankvist o,mirzai b,da silva z,fredlund h,biberfeld g,et al.optimization of polymerase chain reaction for detection of hiv type 2 dna.aids res hum retroviruses.1998;14(13):1151-6.
[0133]
22.de silva ti,cotten m,rowland-jones sl.hiv-2:the forgotten aids virus.trends microbiol.2008;16(12):588-95.
[0134]
23.ferns rb,garson ja.development and evaluation of a real-time rt-pcr assay for quantification of cell-free human immunodeficiency virus type 2 using a brome mosaic virus internal control.j virol methods.2006;135(1):102-8.
[0135]
24.gueudin m,damond f,braun j,taieb a,lemee v,plantier jc,et al.differences in proviral dna load between hiv-1-and hiv-2-infected patients.aids.2008;22(2):211-5.
[0136]
25.chang m,gottlieb gs,dragavon ja,cherne sl,kenney dl,hawes se,et al.validation for clinical use of a novel hiv-2 plasma rna viral load assay using the abbott m2000 platform.j clin virol.2012;55(2):128-33.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。