一种(1s,2s,4s)
‑
β
‑
榄香烯的合成方法及其中间体
技术领域
1.本发明属于药物合成技术领域,具体涉及一种(1s,2s,4s)
‑
β
‑
榄香烯的合成方法及其中间体。
背景技术:
2.随着现代生活环境和饮食习惯的变化,恶性肿瘤已发展成为严重威胁人类生命健康的重大疾病之一。由于肿瘤的传统疗法(如放疗和化疗)毒副作用较大,还容易产生耐药性,寻找和开发新一代高效低毒抗肿瘤药物已成为学术界和工业界共同关注的焦点。其中,来源于自然界且具有良好成药潜力的天然产物便是重要的研究方向之一。
3.榄香烯是一类存在于自然界多种植物挥发油中的倍半萜类天然产物,英文名为elemene,分子式为c
15
h
24
。榄香烯的分子结构中含有3个不饱和双键,根据双键所处位置的差异,可分为α
‑
榄香烯、β
‑
榄香烯、γ
‑
榄香烯和δ
‑
榄香烯。研究表明,榄香烯的这四种同分异构体均具有一定的抗肿瘤活性,但β
‑
榄香烯的药理活性最强,也是目前临床所使用的榄香烯类抗肿瘤药物中最主要的抗肿瘤活性有效成分。目前,β
‑
榄香烯注射液和口服乳这两种剂型已被批准用于胸腹水、食道癌、胃癌、神经胶质瘤和脑转移瘤的临床治疗。与传统的抗肿瘤药物相比,β
‑
榄香烯的最大特点是能透过血脑屏障,毒副作用小且能提高机体的免疫力,同时还能逆转对其他抗肿瘤药物的耐药性。
4.根据4位异丙烯基立体化学的不同,β
‑
榄香烯又分为以下两种非对映异构体:(1s,2s,4r)
‑ꢀ
β
‑
榄香烯和(1s,2s,4s)
‑
β
‑
榄香烯,具体分子结构如下式所示。
[0005][0006]
目前,临床上使用的β
‑
榄香烯注射液和口服乳主要活性有效成分为(1s,2s,4r)
‑
β
‑
榄香烯。但是,研究表明,与(1s,2s,4r)
‑
β
‑
榄香烯相比,(1s,2s,4s)
‑
β
‑
榄香烯具有更高的抗肿瘤活性和更低的毒副作用。
[0007]
目前,(1s,2s,4s)
‑
β
‑
榄香烯主要是从传统中草药莪术(又称“郁金”)中提取分离得到。然而,这天然产物在中药材中含量低,提取分离工艺复杂,且最终产品的纯度不易控制,产量无法保证,极大地限制了β
‑
榄香烯原料药的来源及其临床应用范围。
[0008]
因此,亟需提供一种(1s,2s,4s)
‑
β
‑
榄香烯的合成方法,能够制备出纯度较高,质量易控制的(1s,2s,4s)
‑
β
‑
榄香烯。
技术实现要素:
[0009]
本发明旨在至少解决上述现有技术中存在的技术问题之一。为此,本发明提出一
种 (1s,2s,4s)
‑
β
‑
榄香烯的合成方法,能够制备出纯度大于98%的(1s,2s,4s)
‑
β
‑
榄香烯,且生产效率高。
[0010]
本发明第一方面提供了一种(1s,2s,4s)
‑
β
‑
榄香烯的合成方法。
[0011]
具体的,一种(1s,2s,4s)
‑
β
‑
榄香烯的合成方法,包括以下步骤:
[0012]
(1)在催化剂的作用下,将(r)
‑
香芹酮与异丙烯基格氏试剂进行加成反应,得到 (2r,3r,5r)
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮;
[0013]
(2)在碱液的作用下,将步骤(1)制备的(2r,3r,5r)
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮与含醛基的物质进行烷基化反应,得到(2r,3s,5r)
‑2‑
羟甲基
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮;
[0014]
(3)对步骤(2)制备的(2r,3s,5r)
‑2‑
羟甲基
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮中的羟基进行保护形成保护基,得到化合物a;
[0015]
(4)在还原剂的作用下,将步骤(3)制备的化合物a中酮基还原为羟基,得到化合物 b;
[0016]
(5)将步骤(4)制备的化合物b进行自由基脱氧反应,然后脱去所述保护基,得到 ((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醇;
[0017]
(6)在氧化剂的作用下,将步骤(5)制备的((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑ꢀ
甲醇进行氧化反应,得到((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醛;
[0018]
(7)将正戊基三苯基溴化磷与步骤(6)制备的((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醛进行烯基化反应,得到(1s,2s,4s)
‑
β
‑
榄香烯。
[0019]
优选的,在步骤(1)中,所述催化剂为铜盐。
[0020]
优选的,所述铜盐选自氯化亚铜、溴化亚铜、碘化亚铜、氰化亚铜、三氟甲烷磺酸亚铜、醋酸亚铜、噻吩
‑2‑
甲酸亚铜、四氟硼酸四(乙腈)铜(i)、六氟磷酸四(乙腈)铜(i)或烷基铜(i)盐络合物中的至少一种。
[0021]
优选的,在步骤(1)中,所述异丙烯基格氏试剂为异丙烯基溴化镁格氏试剂。
[0022]
优选的,在步骤(1)中,所述加成反应的反应溶剂选自四氢呋喃、乙醚、乙二醇二甲醚、甲基叔丁基醚、苯或甲苯中的至少一种。
[0023]
优选的,在步骤(2)中,所述含醛基的物质为甲醛和/或多聚甲醛。
[0024]
优选的,在步骤(2)中,所述碱液选自氢氧化钾、氢氧化钠、氢氧化锂、乙醇钠、叔丁醇钾、三乙胺、氢化钾或氢化钠中的至少一种。
[0025]
在步骤(3)中,所述保护基选自酯类保护基、硅醚保护基、苄醚类保护基或烷氧基甲基醚类保护基中的一种。
[0026]
优选的,在步骤(3)中,采用羟基保护剂与步骤(2)制备的(2r,3s,5r)
‑2‑
羟甲基
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮反应,形成所述保护基。
[0027]
优选的,当所述保护基为酯类保护基时,所述羟基保护剂选自酸、酸酐或酰氯中的一种,如乙酸酐、乙酸或乙酰氯。
[0028]
优选的,当所述保护基为硅醚保护基时,所述羟基保护剂选自三甲基氯硅烷(tmscl)、三乙基氯硅烷(tescl)、三异丙基氯硅烷(tipscl)、叔丁基二甲基氯硅烷(tbscl)、叔丁基二苯基氯硅烷(tbdpscl)、三氟甲磺酸三甲基硅酯(tmsotf)、三氟甲磺酸三乙基硅酯
(tesotf)、三氟甲磺酸三异丙基硅酯(tipsotf)、三氟甲磺酸叔丁基二甲基硅酯 (tbsotf)或三氟甲磺酸叔丁基二苯基硅酯(tbdpsotf)中的一种。
[0029]
优选的,当所述保护基为苄醚类保护基,所述羟基保护剂选自溴化苄、对甲氧基溴化苄、或氯化苄。
[0030]
优选的,当所述保护基为烷氧基甲基醚类保护基时,所述羟基保护剂选自氯甲基甲醚 (momcl)或2
‑
(三甲基硅烷基)乙氧甲基氯(semcl)。
[0031]
优选的,在步骤(4)中,所述还原剂选自硼氢化钠、氰基硼氢化钠、硼氢化钾、氢化铝锂、乙硼烷、异丙醇铝或金属钠中的至少一种。所述金属钠在使用中,先溶于乙醇中形成金属钠乙醇溶液。
[0032]
优选的,在步骤(6)中,所述氧化剂选自氯铬酸吡啶嗡盐(pcc)、重铬酸吡啶嗡盐(pdc)、戴斯
‑
马丁氧化剂(dmp)、2
‑
碘酰基苯甲酸(ibx)、二醋酸碘苯/2,2,6,6
‑
四甲基哌啶氧化物 (dib/tempo)、三氧化铬硫酸溶液(jones试剂)、氧化铬
‑
吡啶络合物(sarett试剂或collins 试剂)、二甲亚砜/草酰氯、二甲亚砜/碳二亚胺、二甲亚砜/三氧化硫
‑
吡啶络合物、四丙基高钌酸铵/4
‑
甲基吗啉
‑
n
‑
氧化物(tpap/nmo)、次氯酸盐/2,2,6,6
‑
四甲基哌啶氧化物/溴化物(溴化钠、溴化钾)或二氧化锰中的至少一种。
[0033]
优选的,在步骤(7)中,所述正戊基三苯基溴化磷由正丁基锂和三苯基甲基溴化膦反应制得。
[0034]
优选的,当在步骤(3)中选用醋酸酐作为羟基保护剂,形成酯类保护基时,所述合成方法,包括以下步骤:
[0035]
(1)在催化剂的作用下,将(r)
‑
香芹酮与异丙烯基格氏试剂进行加成反应,得到 (2r,3r,5r)
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮;
[0036]
(2)在碱液的作用下,将步骤(1)制备的(2r,3r,5r)
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮与含醛基的物质进行烷基化反应,得到(2r,3s,5r)
‑2‑
羟甲基
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮;
[0037]
(3)在碱液的作用下,将步骤(2)制备的(2r,3s,5r)
‑2‑
羟甲基
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮与醋酸酐进行酯化反应,得到((1r,4r,6s)
‑1‑
甲基
‑2‑
酮
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯;
[0038]
(4)在还原剂的作用下,将步骤(3)制备的((1r,4r,6s)
‑1‑
甲基
‑2‑
酮
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯进行还原反应,得到((1r,4r,6s)
‑1‑
甲基
‑2‑
羟基
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯;
[0039]
(5)将步骤(4)制备的((1r,4r,6s)
‑1‑
甲基
‑2‑
羟基
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯进行自由基脱氧反应和水解反应,得到((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醇;
[0040]
(6)在氧化剂的作用下,将步骤(5)制备的((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑ꢀ
甲醇进行氧化反应,得到((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醛;
[0041]
(7)将正戊基三苯基溴化磷与步骤(6)制备的((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醛进行烯基化反应,得到(1s,2s,4s)
‑
β
‑
榄香烯。
[0042]
更为具体的,一种(1s,2s,4s)
‑
β
‑
榄香烯的合成方法,包括以下步骤:
[0043]
(1)以铜盐作为催化剂,以(r)
‑
香芹酮为反应的起始原料,与异丙烯基格氏试发生 1,4
‑
迈克尔加成反应,得到(2r,3r,5r)
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮;
[0044]
(2)在热力学条件、碱液的作用下,步骤(1)制备的(2r,3r,5r)
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮与甲醛和/或聚甲醛进行α
‑
烷基化反应,得到(2r,3s,5r)
‑2‑
羟甲基
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮;
[0045]
(3)在碱液的作用下,将步骤(2)制备的(2r,3s,5r)
‑2‑
羟甲基
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮与醋酸酐进行酯化反应,得到((1r,4r,6s)
‑1‑
甲基
‑2‑
酮
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯;
[0046]
(4)在还原剂的作用下,将步骤(3)制备的((1r,4r,6s)
‑1‑
甲基
‑2‑
酮
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯进行还原反应,得到((1r,4r,6s)
‑1‑
甲基
‑2‑
羟基
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯;
[0047]
(5)在经典的巴顿
‑
麦康比自由基脱氧反应条件下,将步骤(4)制备的((1r,4r,6s)
‑1‑ꢀ
甲基
‑2‑
羟基
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯同时进行脱氧反应和水解反应,得到 ((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醇;
[0048]
(6)在氧化剂的作用下,将步骤(5)制备的((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑ꢀ
甲醇进行氧化反应,得到((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醛;
[0049]
(7)先将正丁基锂和三苯基甲基溴化膦反应生成正戊基三苯基溴化磷,然后将所述正戊基三苯基溴化磷与步骤(6)制备的((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醛进行烯基化反应,得到(1s,2s,4s)
‑
β
‑
榄香烯。
[0050]
本发明第二方面提供了合成一种(1s,2s,4s)
‑
β
‑
榄香烯的中间体a。
[0051]
具体的,所述中间体a的结构式如式(a)所示,所述中间体a为((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑ꢀ
二异丙烯基环己烷)
‑1‑
甲醛;
[0052][0053]
本发明第三方面提供了一种合成(1s,2s,4s)
‑
β
‑
榄香烯的中间体b。
[0054]
具体的,所述中间体b的结构式如式(b)所示,所述中间体b为((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑ꢀ
二异丙烯基环己烷)
‑1‑
甲醇;
[0055][0056]
上述中间体产生于(1s,2s,4s)
‑
β
‑
榄香烯的合成过程中,中间体的残留会影响(1s,2s,4s)
‑ꢀ
β
‑
榄香烯的纯度。因此上述中间体能够用于研究(1s,2s,4s)
‑
β
‑
榄香烯的纯度,对控制 (1s,2s,4s)
‑
β
‑
榄香烯的质量具有重要意义。
[0057]
相对于现有技术,本发明的有益效果如下:
[0058]
(1)本发明从起始原料(r)
‑
香芹酮出发,依次经过加成反应、烷基化反应、羟基保护反应、还原反应、自由基脱氧反应、脱去羟基保护基、氧化反应和烯基化反应,首次不对称全合成(1s,2s,4s)
‑
β
‑
榄香烯,其总收率大于8.5%,纯度大于98%(质量分数)。
[0059]
(2)本发明提供的合成方法,反应操作简单,步骤简短,合成效率高;原料(r)
‑
香芹酮便宜易得,生产成本低,适合工业化生产,能够为(1s,2s,4s)
‑
β
‑
榄香烯提供充足来源。
[0060]
(3)本发明提供(1s,2s,4s)
‑
β
‑
榄香烯的中间体,能够用于研究(1s,2s,4s)
‑
β
‑
榄香烯的纯度,对控制(1s,2s,4s)
‑
β
‑
榄香烯的质量具有重要意义。
附图说明
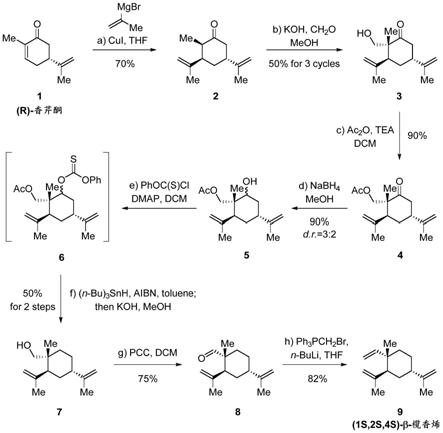
[0061]
图1为实施例1中(1s,2s,4s)
‑
β
‑
榄香烯的合成路线图;
[0062]
图2为实施例1制得的(1s,2s,4s)
‑
β
‑
榄香烯的氢谱图。
具体实施方式
[0063]
为了让本领域技术人员更加清楚明白本发明所述技术方案,现列举以下实施例进行说明。需要指出的是,以下实施例对本发明要求的保护范围不构成限制作用。
[0064]
在以下实施例中,除特别指出,所有反应均在氮气或氩气氛围无水条件下进行;反应使用的试剂均为购买后直接使用(所有化学试剂均从试剂公司购得)。四氢呋喃、甲苯在氮气氛围下由金属钠和二苯乙酮处理;二氯甲烷在氮气氛围下由氢化钙处理。除非特别指明,反应产率均是基于柱色谱的分离产率,柱层析使用硅胶(200
‑
300目)从青岛海洋化工厂购买;反应检测时使用的是0.25mm青岛海洋化工所产的薄层色谱硅胶板(60f
‑
254)。所有核磁共振谱均由br
ü
ker advance 300(1h:300mhz,
13
c:75mhz),br
ü
ker advance 400(1h:400mhz, 13
c:100mhz),br
ü
ker advance 500(1h:500mhz,
13
c:125mhz)仪器测得;如无特殊说明,一般使用氘代氯仿(δh=7.26ppm,δc=77.16ppm)作为溶剂;高分辨质谱均由br
ü
ker apexiv rtms仪器测得;旋光值由horiba sepa
‑
300旋光仪测得。以下是解释多重裂分时所用到的简写:s=单峰,d=双重裂分,t=三重裂分,q=四重裂分,m=多重裂分。
[0065]
实施例1
[0066]
一种(1s,2s,4s)
‑
β
‑
榄香烯的合成方法,包括以下步骤:
[0067]
步骤1:
1h),1.84(d,j=14.3hz,1h),1.76(s,3h),1.71(s,3h),1.05(s,3h).;
13
c nmr(100mhz,cdcl3) δ217.2,146.4,144.4,114.8,112.5,66.5,53.7,42.7,42.1,40.4,28.8,24.0,22.1,16.7;hrms(esi) calcd for c
14
h
23
o2na[m na]
:245.1512;found:245.1512。
[0073]
步骤3:
[0074][0075]
将(2r,3s,5r)
‑2‑
羟甲基
‑2‑
甲基
‑
3,5
‑
二异丙烯基环己酮(4.6g,20.7mmol,1.0equiv)和三乙胺(tea,8.6ml,62.2mmol,3.0equiv)溶解在0℃的二氯甲烷(dcm,100.0ml) 中,然后逐滴加入醋酸酐(ac2o,4.0ml,41.4mmol,2.0equiv),将反应体系缓慢升至室温并搅拌反应过夜,之后使用负载有硅藻土的砂芯漏斗过滤反应混合液,减压浓缩,残余物经快速柱色谱分离(正己烷与乙酸乙酯的体积比由50:1至30:1),以90%的收率得到4.9g 无色透明油状液体((1r,4r,6s)
‑1‑
甲基
‑2‑
酮
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯(式4所示; r
f
=0.75,正己烷:乙酸乙酯=6:1)。((1r,4r,6s)
‑1‑
甲基
‑2‑
酮
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯的1h nmr如下:1h nmr(500mhz,cdcl3)δ4.92
–
4.88(m,1h),4.80(s,1h),4.64(s, 1h),4.57(s,1h),4.21(d,j=10.9hz,1h),3.96(d,j=10.9hz,1h),2.73
–
2.63(m,2h),2.52(t, j=5.6hz,2h),2.02
–
1.95(m,1h),1.94(s,3h),1.85(m,1h),1.68(s,6h),1.01(s,3h);
13
c nmr(125mhz,cdcl3)δ212.5,170.6,146.6,144.0,114.8,111.5,67.1,51.2,44.2,42.4,40.1, 28.9,23.9,21.6,20.7,17.2;hrms(esi)calcd for c
16
h
24
o3na[m na]
:287.1618;found: 287.1615。
[0076]
步骤4:
[0077][0078]
将((1r,4r,6s)
‑1‑
甲基
‑2‑
酮
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯(3.0g,11.3mmol,1.0 equiv)溶解在甲醇(meoh,50.0ml)中,在0℃的条件下加入硼氢化钠(nabh4,0.86g, 22.6mmol,2.0equiv),然后在该温度下搅拌反应1.0小时,最后滴加3n盐酸水溶液(15.0 ml)淬灭反应,用乙酸乙酯(3
×
50.0ml)萃取水相,合并所得有机相,用无水硫酸钠干燥。减压浓缩后,残余物经快速柱色谱分离(洗脱剂为正己烷与乙酸乙酯,洗脱中正己烷与乙酸乙酯的体积比由20:1至15:1),以90%的总收率得到2.7g浅黄色油状液体((1r,4r,6s)
‑1‑
甲基
‑2‑
羟基
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯(式5所示;r
f
=0.40,正己烷:乙酸乙酯=7:1),其是一对难以通过柱色谱实现分离的非对映异构体(d.r.=3:2)。((1r,4r,6s)
‑1‑
甲基
‑2‑
羟基
ꢀ‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯的1h nmr如下:1h nmr(500mhz,cdcl3)δ4.90(s,2h), 4.87(s,0.8h),4.85
–
4.79(m,2.5h),4.73(d,j=9.9hz,2h),4.36(d,j=11.4hz,1h),4.20(d,j =10.9hz,0.7h),4.00(d,j=10.9hz,0.7h),3.76(dd,j=6.5,3.9hz,0.7h),3.55(dd,j=12.0, 3.8hz,2h),2.71(s,1h),2.61
–
2.53(m,0.7h),2.43(s,
1h),2.41
–
2.37(m,0.7h),2.30(dd,j= 13.4,3.0hz,1h),2.06(s,3h),2.04(s,2.3h),2.03
–
1.98(m,1h),1.96
–
1.81(m,3h),1.80(s, 2h),1.77(s,2h),1.73(s,3h),1.71
–
1.67(m,1h),1.03(s,2h),0.84(s,3h);
13
c nmr(125mhz, cdcl3)δ172.2,171.5,149.45,146.47,146.45,145.48,114.1,114.0,110.7,109.5,73.4,69.0,67.4, 67.3,43.5,42.5,41.9,41.3,38.3,38.1,31.6,30.96,29.7,28.6,24.6,23.2,22.8,22.1,21.1,21.0, 18.2,9.8;hrms(esi)calcd for c
16
h
26
o3na[m na]
:289.1774;found:289.1772。
[0079]
步骤5:
[0080][0081]
将((1r,4r,6s)
‑1‑
甲基
‑2‑
羟基
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯(1.5g,5.64mmol,1.0 equiv)溶解在二氯甲烷(dcm,30.0ml)中,之后依次加入4
‑
二甲氨基吡啶(dmap,1.37 g,11.28mmol,2.0equiv)、硫代氯甲酸苯酯(phoc(s)cl,1.94g,11.28mmol,2.0equiv),在室温条件下搅拌反应过夜,然后使用负载有硅藻土的砂芯漏斗过滤反应混合液,减压浓缩得到((1r,4r,6s)
‑1‑
甲基
‑2‑
硫代甲酸苯酯
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯(式6所示)的反应混合物。在氩气氛围下将((1r,4r,6s)
‑1‑
甲基
‑2‑
硫代甲酸苯酯
‑
4,6
‑
二异丙烯基环己烷)
‑1‑
乙酸甲酯直接溶解于甲苯(meoh,20.0ml)中,加热至回流状态后加入三丁基氢化锡((n
‑
bu)3snh, 3.30g,11.28mmol,2.0equiv)和自由基引发剂偶氮二异丁腈(aibn,370mg,11.28mmol, 0.2equiv),在该温度下搅拌反应8.0小时,随后将反应体系温度降至室温,并加入溶解在甲醇(toluene,30.0ml)的氢氧化钾(koh,1.0g),继续搅拌反应2.0小时,最后滴加饱和氯化铵水溶液(10.0ml),用乙酸乙酯(3
×
30ml)萃取水相,合并所得有机相,用无水硫酸钠干燥。减压浓缩后,残余物经快速柱色谱分离(洗脱剂为正己烷与乙酸乙酯,洗脱中正己烷与乙酸乙酯的体积比由=50:1至20:1),以两步50%的总收率得到580mg白色固体 ((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醇(式7所示;r
f
=0.30,正己烷:乙酸乙酯=10:1)。((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醇的1h nmr如下:1h nmr(500 mhz,cdcl3)δ4.88(d,j=1.3hz,1h),4.82(s,1h),4.79(s,1h),4.76(d,j=1.5hz,1h),3.39
–ꢀ
3.30(m,2h),2.35(s,1h),2.28(dd,j=10.9,3.7hz,1h),1.81(m,2h),1.76(s,3h),1.72(s,3h), 1.71
–
1.62(m,3h),1.56
–
1.48(m,1h),1.17(dd,j=9.1,4.1hz,1h),0.94(s,3h);
13
c nmr (125mhz,cdcl3)δ149.2,147.2,112.9,110.5,72.3,44.8,39.0,38.9,31.8,29.9,23.9,23.3,22.5, 17.9;hrms(esi)calcd for c
14
h
25
o[m h]
:209.1900;found:209.1900。
[0082]
步骤6:
[0083]
[0084]
将((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醇(410mg,1.97mmol,1.0equiv) 溶解在二氯甲烷(20.0ml)中,在0℃的条件下加入氯铬酸吡啶嗡盐(847mg,3.94mmol, 2.0equiv)和柱层析用硅胶(200
‑
300目,800mg),之后将反应缓慢升至室温并搅拌反应3.0 小时。将反应混合液直接减压浓缩后,残余物经快速柱色谱分离(洗脱剂为正己烷与乙酸乙酯,洗脱中正己烷与乙酸乙酯的体积比由80:1至50:1),以75%的收率得到314mg浅黄色油状液体((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醛(式8所示;r
f
=0.60,正己烷:乙酸乙酯=10:1)。((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醛的1h nmr如下:1h nmr(400mhz,cdcl3)δ9.39(s,1h),4.91
–
4.88(m,1h),4.82(d,j=1.2hz,1h),4.77(s,1h), 4.74(s,1h),2.67(dd,j=7.2,4.8hz,1h),2.38
–
2.28(m,1h),1.93
–
1.84(m,1h),1.74(s,3h), 1.70(s,3h),1.68
–
1.70(m,4h),1.39(m,1h),1.00(s,3h);
13
c nmr(100mhz,cdcl3)δ206.4, 147.9,146.3,113.6,110.1,49.5,42.5,38.5,30.5,29.4,25.4,24.9,21.8,17.1;hrms(esi)calcd for c
14
h
23
o[m h]
:207.1743;found:207.1742。
[0085]
步骤7:
[0086][0087]
在氩气的氛围下将三苯基甲基溴化膦(571mg,1.60mmol,2.2equiv)溶解在四氢呋喃 (10.0ml)中,然后在
‑
78℃的条件下逐滴加入正丁基锂溶液(1.6m,0.9ml,1.46mmol, 2.0equiv),并在该温度下继续搅拌反应30分钟后缓慢升至室温,搅拌反应20分钟,制得正戊基三苯基溴化磷。随后将反应体系重新置于
‑
78℃的条件,逐滴加入溶解于四氢呋喃(2.0 ml)的((1r,2s,4s)
‑1‑
甲基
‑
2,4
‑
二异丙烯基环己烷)
‑1‑
甲醛(150mg,0.73mmol,1.0equiv),并在该温度下继续搅拌反应30分钟后缓慢升至室温,搅拌反应30分钟,最后滴加饱和氯化铵水溶液(5.0ml)淬灭反应,用乙酸乙酯(3
×
20ml)萃取水相,合并所得有机相,用无水硫酸钠干燥。减压浓缩后,残余物经快速柱色谱分离(洗脱剂为100%正己烷),以82%的收率得到121mg无色透明液体(1s,2s,4s)
‑
β
‑
榄香烯(式9所示;r
f
=0.95,100%正己烷)。 (1s,2s,4s)
‑
β
‑
榄香烯的旋光值:(1s,2s,4s)
‑
β
‑
榄香烯的1h nmr如下:1h nmr(500mhz,cdcl3)δ5.82(dd,j=17.5,11.0hz,1h),4.93(dd,j=8.3,1.0hz,1h), 4.90(s,1h),4.86(d,j=1.2hz,1h),4.84(m,1h),4.79(s,1h),4.68(s,1h),2.40
–
2.32(m,1h), 2.18(m,1h),1.75(s,3h),1.73(s,3h),1.57
‑
1.78(m,5h),1.32(m,1h),1.02(s,3h);
13
c nmr (125mhz,cdcl3)δ150.2,148.2,147.9,112.4,110.17,110.15,47.7,39.8,39.1,34.7,30.2,25.5, 24.6,22.30,22.29;hrms(esi)calcd for c
15
h
25
[m h]
:205.1951;found:205.1952((1s,2s,4s)
‑ꢀ
β
‑
榄香烯的氢谱图参见图2)。经高效液相色谱(hplc)和气相色谱(gc)分析,所合成的(1s,2s,4s)
‑
β
‑
榄香烯的纯度为98.89%。
[0088]
以上步骤1
‑
7的合成路线图参见图1。步骤1
‑
6合成了(1s,2s,4s)
‑
β
‑
榄香烯的中间体a,步骤1
‑
5合成了(1s,2s,4s)
‑
β
‑
榄香烯的中间体b。
[0089]
经过多次试验,按照本发明提供的合成方法制备的(1s,2s,4s)
‑
β
‑
榄香烯的纯度
大于98.0%,能够满足药理学、药效学等成药性的研究。与(1s,2s,4r)
‑
β
‑
榄香烯相比,(1s,2s,4s)
‑ꢀ
β
‑
榄香烯具有更高的抗肿瘤活性和更低的毒副作用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。