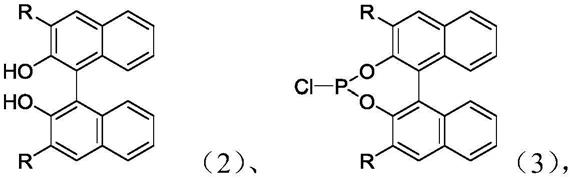
1.本发明涉及一种磷酸酯配体及其制备方法,还涉及其在端烯烃氢甲酰化制备线性醛中的应用。
背景技术:
2.氢甲酰化是一种以烯烃为原料来制备多一个碳原子醛的合成方法,在工业化生产中有着巨大的应用。所制备的醛通过加氢/氧化/胺化成脂族醇/脂族酸/脂族胺,常用于聚酯单体、以及增塑剂。目前氢甲酰化反应的主要类型是使用与磷配体络合的viii族过渡金属,烯烃化合物与一氧化碳和氢的加氢甲酰化。
3.在氢甲酰化反应中,诸如产物选择性、催化剂反应性以及催化剂和配体稳定性等因素常常是选择所用磷配体的主要关注问题。专利us5235113利用多齿亚磷酸盐四(二
‑
(2,4
‑
二
‑
叔丁基苯基)磷)
‑
季戊四醇为配体得到末端醛的选择性仅为30%;专利cn97194561.6提供了一种二齿有机亚磷酸盐配位体用于催化单烯烃氢甲酰化,得到端醛的选择性也仅为70%。
4.此外,现有技术中采用的这些磷酸酯配体还存.//在水解问题,磷酸酯水解后形成的小分子膦有机物会进一步催化副产物缩醛的产生,对后续精馏产品纯度会产生关键影响。
5.因此,亟需研发一种新的催化剂含磷配体,来改善氢甲酰化的选择性。
技术实现要素:
6.为克服现有技术中存在的上述缺陷,本发明的目的是提供一种用于端烯烃氢甲酰化反应中的磷酸酯配体,所述配体水解稳定性好,反应活性高。
7.本发明的另一个目的在于提供所述磷酸酯配体的制备方法。
8.本发明的再一目的是提供一种所述配体在催化端烯烃氢甲酰化制备线性醛中的应用,具有高反应活性和线性选择性。
9.为实现上述目的,本发明采用的技术方案如下:
10.本发明提供一种磷酸酯配体,其结构如式1所示:
[0011][0012]
式中:r为c1‑
c6烷基、苯基、噻吩基,优选为乙烷基、苯基。
[0013]
本发明还提供了一种上述式1所示磷酸酯配体的制备方法,步骤包括:
[0014]
1)式2所示单体与三氯化磷(pcl3)在三乙胺(net3)存在下发生加成反应,生成式3所示中间体;
[0015]
2)式3所示中间体与外
‑
内
‑
双环[2.2.1]庚烷
‑
2,6
‑
二醇在三乙胺(net3)存在下发生取代反应,生成式1所示磷酸酯配体;
[0016]
步骤1)中所述式2、步骤2)中所述式3结构如下:
[0017][0018]
式2、式3中r定义与式1中相同,即r为c1‑
c6烷基、苯基、噻吩基,优选为乙烷基、苯基;即式2所示单体更优选为3,3'
‑
二苯基
‑
2,2'
‑
二羟基
‑
1,1'
‑
二萘基(cas号:102490
‑
05
‑
1,75640
‑
70
‑
9,75684
‑
93
‑
4)、3,3'
‑
二乙基
‑
2,2'
‑
联萘
‑
2,2'
‑
二醇(cas号:957230
‑
60
‑
3)。
[0019]
步骤2)中所述外
‑
内
‑
双环[2.2.1]庚烷
‑
2,6
‑
二醇(cas号:14339
‑
82
‑
3)结构如式4所示:
[0020][0021]
本发明所述式1所示磷酸酯配体的制备方法用反应式表示如下:
[0022][0023]
本发明制备方法,步骤1)中,所述三氯化磷的用量为式2所示单体摩尔量的1.0
‑
5.0倍例如1.5倍、2.0倍、2.5倍、3.0倍、3.5倍、4.0倍、4.5倍,优选1.0
‑
2.0倍;
[0024]
所述三乙胺的用量为式2所示单体摩尔量的0.5
‑
5.0倍例如1.0倍、1.5倍、2.0倍、2.5倍、3.0倍、3.5倍、4.0倍、4.5倍,优选2.0
‑
3.0倍。
[0025]
本发明制备方法,步骤1)中,所述加成反应在溶剂存在下进行,所述溶剂为甲苯、四氢呋喃、氯苯中的任意一种或至少两种的组合,优选为四氢呋喃;
[0026]
所述溶剂用量没有具体要求,能够完全溶解反应原料,使加成反应顺利进行即可;优选地,所述溶剂用量为式2单体摩尔量的1.0
‑
5.0倍例如1.5倍、2.0倍、2.5倍、3.0倍、3.5倍、4.0倍、4.5倍,优选3.0
‑
5.0倍。
[0027]
本发明制备方法,步骤1)中,所述加成反应,反应温度为室温下反应例如25℃、28℃、30℃、35℃,优选25
‑
30℃;反应时间为1.0
‑
5.0h例如1.2h、1.5h、2.0h、2.5倍、3.0h、3.5h、4.0h、4.5h,优选1.5
‑
2.5h。
[0028]
本发明制备方法,步骤2)中,所述外
‑
内
‑
双环[2.2.1]庚烷
‑
2,6
‑
二醇的用量为式3所示中间体摩尔量的0.1
‑
1.0倍例如0.2倍、0.3倍、0.4倍、0.5倍、0.6倍、0.7倍、0.8倍、0.9倍,优选0.3
‑
0.5倍;
[0029]
所述三乙胺的用量为式3所示中间体摩尔量的10
‑
30倍例如12倍、15倍、18倍、20倍、25倍、28倍,优选15
‑
20倍。
[0030]
本发明制备方法,步骤2)中,所述取代反应在溶剂存在下进行,所述溶剂为甲苯、四氢呋喃、氯苯中的一种或多种,优选采用与步骤1)同种类的溶剂,更优选为四氢呋喃;
[0031]
所述溶剂用量没有具体要求,能够完全溶解反应原料,使加成反应顺利进行即可;优选地,所述溶剂用量为三乙胺摩尔量的0.05
‑
0.2倍例如0.06倍、0.08倍、0.09倍、0.10倍、0.12倍、0.14倍、0.16倍、0.18倍,优选0.08
‑
0.1。
[0032]
本发明制备方法,步骤2)中,所述取代反应,反应温度为室温下反应例如25℃、28℃、30℃、35℃,优选25
‑
30℃;反应时间为0.5
‑
5.0h例如1.2h、1.5h、2.0h、2.5倍、3.0h、3.5h、4.0h、4.5h,优选1.0
‑
2.0h。
[0033]
本发明制备方法,在步骤1)反应完成后,可以采用蒸馏、精馏等常规后处理方法,将式3所示中间体精制后使用,也可以在加成反应完成后,不经后处理,将反应体系直接套用到步骤2)中使用;优选不经后处理直接套用。
[0034]
作为优选,在本发明一些示例中,具体采用的制备方法,步骤为:将式2所示单体、三氯化磷、三乙胺溶于四氢呋喃中,室温下反应得到中间体;然后向反应体系中加入外
‑
内
‑
双环[2.2.1]庚烷
‑
2,6
‑
二醇,并补充三乙胺、四氢呋喃至所需用量,室温下反应得到磷酸酯配体。
[0035]
本发明所述的磷酸酯配体可用于催化烯烃氢甲酰化反应,尤其适用于催化端烯烃氢甲酰化制备相应的线性醛。
[0036]
适用本发明磷酸酯配体的所述端烯烃为c3‑
c
10
的端烯烃,优选为1
‑
丁烯、丙烯、1,3
‑
丁二烯、1,4
‑
戊二烯、1,5
‑
己二烯、1,6
‑
庚二烯。
[0037]
作为优选,本发明提供一种端烯烃氢甲酰化制备线性醛的方法,步骤包括:
[0038]
将催化剂、所述磷酸酯配体溶解于溶剂中,随后加入端烯烃,并通入合成气,升温至反应温度进行氢甲酰化反应,反应过程通过合成气控制反应压力,反应一定时间得到线性醛;
[0039]
本发明所述催化剂选自过渡金属化合物,所述过渡金属化合物选自醋酸铑、辛酸铑、乙酰丙酮铑、乙酰丙酮羰基铑、二羰基乙酰丙酮铑、三苯基膦乙酰丙酮铑、醋酸钴、辛酸钴、乙酰丙酮钴、乙酰丙酮羰基钴、三苯基膦乙酰丙酮钴中的任意一种或至少两种的组合,优选为二羰基乙酰丙酮铑和/或三苯基膦乙酰丙酮钴。
[0040]
本发明所述磷酸酯配体的加入量为催化剂摩尔量的30
‑
100倍例如35倍、40倍、50倍、60倍、70倍、80倍、90倍,优选40
‑
60倍。
[0041]
本发明所述催化剂的加入量为端烯烃摩尔量的0.01
‑
0.2倍例如0.02倍、0.04倍、0.05倍、0.08倍、0.10倍、0.15倍,优选0.01
‑
0.05倍。
[0042]
本发明所述合成气,其中co/h2摩尔比为1:0.8
‑
1.2例如1:0.9、1:1、1:1.2,进料量通过反应压力调节。
[0043]
本发明所述溶剂选自四氢呋喃、二氯甲烷、苯、甲苯、氯仿、正己烷中的任意一种或
至少两种的组合,优选为苯和/或甲苯;
[0044]
优选地,所述溶剂用量为烯烃摩尔量的2.0
‑
4.0例如2.0倍、2.5倍、3.0倍、3.5倍、4.0倍,优选2.0
‑
3.0。
[0045]
本发明所述氢甲酰化反应,反应压力为1.0
‑
10.0mpag例如1.0mpag、3.0mpag、5.0mpag、7.0mpag、9.0mpag,优选1.0
‑
5.0mpag;反应温度为50
‑
100℃例如55℃、60℃、65℃、70℃、80℃、90℃,优选55
‑
70℃,反应时间为1.0
‑
5.0h例如1.2h、1.5h、2.0h、2.5倍、3.0h、3.5h、4.0h、4.5h,优选1.0
‑
2.5h。
[0046]
本发明所述氢甲酰化反应,转化率可达95%以上,选择性达96%以上,二缩醛选择性降低至0.5%以下。
[0047]
本发明氢甲酰化反应后体系通过简单精馏即可得到高纯度线性醛产品,所述精馏为本领域常规操作,由此得到的精馏产品纯度可高达99.9%以上。
[0048]
本发明所述磷酸酯配体,具有非极性的大共轭碳环、桥环结构,可将亚磷酸酯基团包裹起来,使水分子难以与亚磷酸酯基团接触,因此具有水解稳定性好,反应活性高的优点。
[0049]
将本发明所述磷酸酯配体用于催化端烯烃氢甲酰化制备线性醛,一方面是利用萘环大位阻使氢甲酰化反应倾向于在端基加成;另一方面磷酸酯
‑
外
‑
内
‑
双环[2.2.1]庚烷
‑
2,6
‑
二醇的刚性桥碳结构能够使p
‑
p
‑
金属之间的配位空间保持在适宜的范围内,不会因空间过大而降低反应效率;此外在富电子的磷酸酯中氧的强吸电子使得在配位时m
‑
co键减弱,易于co转移到烯烃双键上,有利于加快反应速率,降低反应温度,减少副产物的生成。
[0050]
与现有技术相比,本发明有益效果在于:
[0051]
本发明提供的磷酸酯配体,与金属有较强的鳌合能力,金属活性中心稳定性好,耐水解,催化活性高。使用该配体催化端烯烃氢甲酰化制备线性醛,能够提高低温反应速率,线性醛选择性好(选择性可以达90~95%),与金属有较强的螯合能力,反应活性高并且能够减少副产物缩醛的产生,后续精馏产品纯度高。本发明制备线性醛的方法具有工艺简便、成本与能耗低、生产安全性好、设备投资低,所得产品质量高等多重优点,特别适用于大规模的工业化生产。
具体实施方式
[0052]
以下结合具体实施例对本发明的技术方案做进一步详细说明。
[0053]
本发明实施例和对比例中使用的试剂原料来源如下:
[0054]
外
‑
内
‑
双环[2.2.1]庚烷
‑
2,6
‑
二醇、二羰基乙酰丙酮铑、三苯基膦乙酰丙酮钴、(r)
‑
3,3'
‑
二乙基
‑
1,1'
‑
联萘
‑
2,2'
‑
二醇、(s)
‑
3,3'
‑
二苯基
‑
2,2'
‑
二羟基
‑
1,1'
‑
二萘基、购自sigma aldrich试剂公司;
[0055]
三乙胺、三氯化磷、甲苯、四氢呋喃购自上海国药试剂有限公司;
[0056]
其余试剂原料如无特别说明,均为市售产品。
[0057]
本发明实施例和对比例中使用的测试方法如下:
[0058]
产物结构由元素分析仪器测定,仪器为德国elementar公司vario el cube分析仪。
[0059]
产品含量分析方法:agilent 7890b气相色谱仪,安捷伦db
‑
5色谱柱,进样口温度:
220℃;检测器温度:250℃;h2流量:40/min;空气流量:360ml/min。柱箱升温程序为:初始温度20℃,升温速率为20℃/min,保持4min;100
‑
250℃,升温速率15℃/min,保持10min。
[0060]
产品结构分析:核磁bruker avanceiii 500mhz分析仪。
[0061]
以下结合具体实施例,对本发明作进一步说明。应理解,以下实施例仅用于说明本发明而非用于限定本发明的范围。
[0062]
实施例1
[0063]
(1)制备磷酸酯配体,步骤为:
[0064]
1)将(r)
‑
3,3'
‑
二乙基
‑
1,1'
‑
联萘
‑
2,2'
‑
二醇(3424.4g,10mol)、三乙胺(3035.7g,30mol)、三氯化磷(2746.6,20mol)溶于3.5l四氢呋喃(43.11mol)中,室温30℃下反应2.5小时,得到中间体(3498.9g,8.6mol),结构如下:
[0065][0066]
核磁分析:1h nmr(500mhz,chloroform
‑
d)δ7.85(dt,2h),7.70(d,2h),7.46(t,2h),7.39(d,2h),7.28(ddd,2h),2.66(d,4h),1.26(t,6h)
[0067]
2)随后将外
‑
内
‑
双环[2.2.1]庚烷
‑
2,6
‑
二醇(542.5g,4.3mol)、三乙胺(17.4kg,172.0mol)溶于2.8l四氢呋喃(34.4mol)中,加入到步骤1)反应液中混合均匀,室温30℃反应2.0h,得到磷酸酯配体(3475.8g,4.0mol),结构如下:
[0068][0069]
元素分析:c:76.01;h:5.85;p:7.18;o:10.96。
[0070]
核磁分析:1h nmr(500mhz,chloroform
‑
d)δ7.85(dt,4h),7.67(d,4h),7.49
‑
7.40(t,8h),7.29(d,4h),4.41(m,2h),2.65(m,8h),2.50(m,1h),2.22(m,1h),1.87(dd,2h),1.72
‑
1.64(d,2h),1.55(dd,2h),1.33(t,12h)。
[0071]
(2)1,3
‑
丁二烯氢甲酰化制备1,5
‑
已二醛
[0072]
将三苯基膦乙酰丙酮钴、上述步骤(1)制备的磷酸酯配体溶于甲苯溶剂中,随后加入1,3
‑
丁二烯混合均匀,1,3
‑
丁二烯、三苯基膦乙酰丙酮钴、配体、甲苯摩尔比为1:0.01:0.4:2.0,加入到反应釜中,并通入合成气(co/h2摩尔比为1:1)控制体系压力为1.0mpag,在合成气氛围、温度55℃下反应1.0h,由气相色谱分析得到直链产物1,5
‑
已二醛;1,3
‑
丁二烯转化率为99.3%,1,5
‑
已二醛选择性为96.7%,副产二缩醛选择性为0.5%。
[0073]
反应液放置24h后,检测其中水含量为0.35wt%,测试配体含量较反应前配体初始
加入量降低为99.5%,磷酸酯配体水解率为0.5%。
[0074]
反应液精馏收集1,5
‑
已二醛产品,纯度为99.91%。
[0075]
实施例2
[0076]
(1)制备磷酸酯配体,步骤为:
[0077]
1)将(s)
‑
3,3'
‑
二苯基
‑
2,2'
‑
二羟基
‑
1,1'
‑
二萘基(4385.3g,10mol)、三乙胺(3035.7g,30mol)、三氯化磷(2746.6,20mol)溶于3.5l四氢呋喃(43.11mol)中,室温30℃下反应2.5小时,得到中间体(4325.2g,8.6mol),结构如下:
[0078][0079]
核磁分析:1h nmr(500mhz,chloroform
‑
d)δ8.01(m,4h),7.62(m,16h)。
[0080]
2)随后将外
‑
内
‑
双环[2.2.1]庚烷
‑
2,6
‑
二醇(542.5g,4.3mol)、三乙胺(17.4kg,172.0mol)溶于2.8l四氢呋喃(34.4mol)中,加入到步骤1)反应液中混合均匀,室温30℃反应2.0h,得到磷酸酯配体(3475.8g,4.0mol),结构如下:
[0081][0082]
元素分析:c:80.35;h:4.78;p:5.79;o:9.08。
[0083]
核磁分析:1h nmr(500mhz,chloroform
‑
d)δ8.09
–
7.99(m,8h),7.50
–
7.34(m,30h),4.45(dq,2h),2.48(m,2h),2.20(m,2h),1.87(dt,2h),1.72
–
1.64(m,2h),1.55(t,2h)。
[0084]
(2)1,4
‑
戊二烯氢甲酰化制备1,6
‑
庚二醛
[0085]
将二羰基乙酰丙酮铑、上述步骤(1)制备的磷酸酯配体溶于甲苯溶剂中,随后加入1,4
‑
戊二烯混合均匀,1,4
‑
戊二烯、二羰基乙酰丙酮铑、配体、甲苯摩尔比为1:0.05:3.0:2.5,加入到反应釜中,并通入合成气(co/h2摩尔比为1:1)控制体系压力为5.0mpag,在合成气氛围、温度60℃下反应2.5h,由气相色谱分析得到直链产物1,6
‑
庚二醛;1,4
‑
戊二烯转化率为98.7%,1,6
‑
庚二醛选择性为95.1%,副产二缩醛选择性为0.42%。
[0086]
反应液放置24h后,检测其中水含量为0.37wt%,测试配体含量较反应前配体初始加入量降低为99.68%,磷酸酯配体水解率为0.32%。
[0087]
反应液采用精馏提纯收集1,6
‑
庚二醛产品,纯度为99.95%。
[0088]
实施例3
[0089]
(1)配体耐水解性能测试
[0090]
采用实施例1方法由1,3
‑
丁二烯氢甲酰化制备1,5
‑
已二醛,反应完成后反应液通过精馏将产品脱出,溶剂及配体循环反应30次,测试其中水含量和配体含量,计算配体含量较反应前配体初始加入量减少百分率,即配体水解率,结果如下:
[0091]
循环次数水含量(%)配体水解率(%)50.430.4100.550.6150.620.9200.711.0250.821.2300.961.3
[0092]
对比例1
[0093]
1,3
‑
丁二烯氢甲酰化制备1,5
‑
已二醛:
[0094]
参照实施例1氢甲酰化反应,不同之处仅在于:将本发明磷酸酯配体替换为同摩尔量的三苯基亚磷酸酯配体,其它操作步骤及反应条件与实施例1相同。
[0095]
由气相色谱分析得到直链产物1,5
‑
已二醛;1,3
‑
丁二烯转化率为72.7%,1,5
‑
已二醛选择性为68.8%,副产二缩醛选择性为1.5%。
[0096]
反应液放置24h后,检测其中水含量为0.51%,测试配体含量较反应前配体初始加入量降低为95.6%,三苯基亚磷酸酯配体水解率为4.4%。
[0097]
反应液采用精馏提纯收集壬醛产品,纯度为93.2%。
[0098]
对比例2
[0099]
1,3
‑
丁二烯氢甲酰化制备1,5
‑
已二醛:
[0100]
参照实施例1氢甲酰化反应,不同之处仅在于:将本发明磷酸酯配体替换为同摩尔量的步骤1制备的中间体,其它操作步骤及反应条件与实施例1相同。
[0101]
由气相色谱分析得到直链产物1,5
‑
已二醛;1,3
‑
丁二烯转化率为52.7%,1,5
‑
已二醛选择性为62.0%,副产二缩醛选择性为3.5%。
[0102]
反应液放置24h后,检测其中水含量为0.43%,测试配体含量较反应前配体初始加入量降低为96.1%,三苯基亚磷酸酯配体水解率为3.9%。
[0103]
反应液采用精馏提纯收集壬醛产品,纯度为89.6%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。