1.本发明属于催化材料技术领域,具体涉及一种异相结锰氧化物催化剂及其制备方法与应用。
背景技术:
2.甲醛作为室内最严重的空气污染物之一,对人体健康的危害最为突出。因其具有与体内蛋白质等生物质发生反应而引起生物质变性的高化学反应活性,长期暴露在浓度超标的甲醛环境中可能会引发鼻膜炎、咽喉炎、支气管炎、哮喘等一系列呼吸道疾病,严重情况下甚至导致癌变、白血病。因此,研究开发安全高效的甲醛净化消除技术具有重要意义。与其他净化技术相比,催化氧化法以其环境友好性、设备简单、无次生污染物和普适性强等优点,被认为是最具前景的甲醛消除技术。发展安全高效的甲醛氧化催化剂是推进催化氧化消除甲醛技术实际应用的关键。
3.催化氧化技术的关键是催化剂的选取,根据催化剂主体材料的不同,可分为负载型贵金属催化剂和非贵金属催化剂(过渡金属催化剂)。以pt,au,pd和ag等为代表的负载型贵金属催化剂具备优异的反应活性,能在低温甚至室温下实现甲醛的完全氧化。然而贵金属的价高稀缺限制了该类催化剂的广泛应用,如何发展一种高效、廉价的催化剂成为了研究重点。因此以过渡金属催化剂(mno2、ceo2、co3o4等)为代表的非贵金属催化剂受到了广泛关注。近年来,设计提高非贵金属催化剂的催化活性以获得具有应用价值的催化剂材料已成为发展甲醛催化氧化技术的主流趋势。根据文献报道,对非贵金属催化剂的改性主要集中于组分调制和结构优化,常采用掺杂和复合以及结构纳米化、形貌控制和缺陷调变等策略。但总体而言,非贵金属催化剂仍普遍存在活性偏低、长期工作稳定性和耐湿性欠佳等问题,因此发展高活性的非贵金属催化剂设计理念与可控合成方法仍是推进甲醛催化氧化技术实用化进程中亟待解决的关键问题。
技术实现要素:
4.针对以上现有技术存在的缺点和不足,本发明的首要目的在于提供一种异相结锰氧化物催化剂。本发明的催化剂具有大量异相结相界面和富含氧空位的特征,兼具高本征催化活性和丰富的活性位点。
5.本发明的另一目的在于提供上述异相结锰氧化物催化剂的制备方法。本发明采用水热结合球磨处理两步法。首先以含有高锰酸钾和锰盐的水溶液为起始原料,采用水热方法制备得到亚稳相δ
‑
mno2前躯体,然后经球磨诱导产生相变(δ
‑
mno2转变为α
‑
mno2,该相变过程经历非晶中间相),选择合适的球磨条件得到具备同质异相结的锰氧化物催化剂。该方法原料易得、操作简便、便于量产,使得工业化生产成为可能。
6.本发明的再一目的在于提供上述异相结锰氧化物催化剂在甲醛催化氧化中的应用,可在80℃下高效且稳定地催化甲醛氧化分解,其活性优于多数目前报道的锰基非贵金属催化剂。
7.本发明目的通过以下技术方案实现。
8.一种异相结锰氧化物催化剂,所述异相结锰氧化物催化剂由非晶相锰氧化物和α
‑
mno2组成,所述非晶相锰氧化物和α
‑
mno2的相界面为同质异相结。
9.优选地,所述锰氧化物是指具有多晶型现象的非贵金属氧化物mno2。
10.优选的,所述同质异相结界面富含氧空位。
11.以上任一项所述的一种异相结锰氧化物催化剂的制备方法,包括如下步骤:
12.将mno2在空气氛围下球磨,制得异相结锰氧化物催化剂;所述mno2为δ
‑
mno2。
13.优选的,所述球磨的时间为0.5~20h。
14.优选的,所述球磨的转速为50~1200rpm。
15.优选的,所述球磨的球料比为10:1~200:1。
16.优选的,所述球磨的时间为3h;所述球磨的转速为600rpm;所述球磨的球料比为100:1。
17.优选的,所述球磨过程中,球磨机为行星式球磨机、摆振球磨机、振动式等离子球磨机中的至少一种,球磨罐和研磨球为不锈钢、玛瑙、氧化锆、聚四氟乙烯材质中至少一种。
18.优选的,所述δ
‑
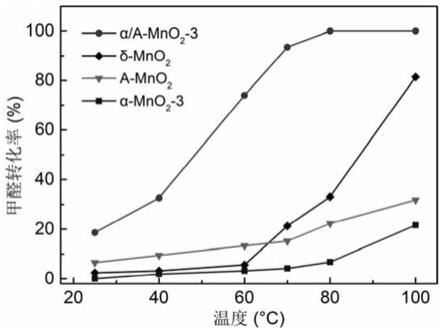
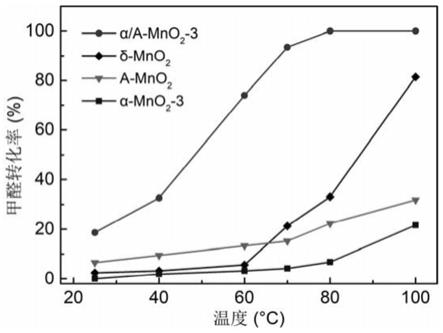
mno2的制备包括以下步骤:
19.将高锰酸钾、锰盐溶于水中,经超声、搅拌后水热反应,反应完成后冷却至室温,所得沉淀物经清洗、干燥后制得δ
‑
mno2。
20.优选的,所述锰盐是指mnso4、mn(no3)2、mnco3、mn(ch3coo)2中至少一种;所述超声的时间为1~2小时;所述搅拌的时间为0.5~2小时;所述清洗是指分别用超纯水、无水乙醇清洗;所述高锰酸钾加入到水中的浓度为0.012~0.03m,锰盐加入到水中的浓度为0.002~0.005m;所述水热反应的温度为140~180℃,时间为8~16h。
21.优选地,所述水热反应温度为160℃;时间为6~12h。
22.以上任一项所述的一种异相结锰氧化物催化剂在甲醛催化氧化中的应用。
23.本发明的原理为:对于甲醛催化氧化催化剂,催化剂对甲醛分子的吸附能力和对氧分子的活化能力是影响催化性能的关键。目前的非贵金属催化剂往往因吸附甲醛分子能力较弱或难以持续产生活性氧而难以实现甲醛的低温催化氧化。本发明所提供的催化剂在设计思路上引入异相结的协同作用来优化甲醛分子的吸附和活性氧产生两个方面,并提供了简单易行的制备方法加以实现。本发明采用水热结合球磨两步法合成了具备同质异相结的锰氧化物催化剂。首先通过水热法得到容易产生相变的亚稳相前躯体δ
‑
mno2;随后,通过调控球磨条件来得到具备同质异相结的催化剂。所得非晶锰氧化物和alpha相二氧化锰组合构筑协同催化活性位,其中非晶锰氧化物相提供分解氧分子产生活性氧物种的活性位,alpha相二氧化锰提供甲醛吸附位点。在本发明中,异相结界面的晶格高度不匹配使其具有丰富的缺陷结构;此外,机械作用可进一步提高催化剂的氧缺陷含量。非晶锰氧化物和alpha相二氧化锰组合构筑同质异相结结合丰富的氧空位,为甲醛催化氧化提供了大量的活性位点与吸附位点,协同地提高了甲醛催化氧化的活性。综上,本发明所提供的甲醛催化氧化催化剂兼具高本征活性和丰富的活性位。
24.相对于现有技术,本发明具有如下优点及有益效果:
25.(1)本发明提供的方法与材料有效的兼具优化本征活性和活性位数量。通过构筑同质异相结形成大量相界面有效引入大量的氧空位,所得非晶锰氧化物和alpha相二氧化
锰组合构筑协同催化活性位,有效提高催化剂活性和催化稳定性。
26.(2)本发明的制备方法原料易得、工艺简单、便于量产、使得工业化生产成为可能。
27.(3)本发明所得锰氧化物催化剂可在80℃下实现甲醛完全氧化去除且其具有较高的比表面积反应速率(高达0.21μmol m
‑2min
‑1);此外,其表现出优异的稳定性,其活性优于多数目前报道的锰基非贵金属催化剂。
附图说明
28.图1为本发明实施例1中前躯体δ
‑
mno2,目标催化剂α/a
‑
mno2‑
3和参比样品α
‑
mno2‑
3和a
‑
mno2的x射线衍射图;其中,3指球磨时间(h)。
29.图2为本发明实施例1中所得目标催化剂α/a
‑
mno2‑
3的透射电镜形貌图(a);选区电子衍射图谱(b)和高分辨透射电子显微照片(c)。
30.图3a为本发明实施例1中所得目标催化剂α/a
‑
mno2‑
3在o1s区域的的x射线光电子能谱图。
31.图3b为本发明实施例1中所得目标催化剂α/a
‑
mno2‑
3在mn 2p区域的的x射线光电子能谱图。
32.图3c为本发明实施例1中所得目标催化剂α/a
‑
mno2‑
3在mn 3s区域的的x射线光电子能谱图。
33.图4为本发明实施例1中所得前躯体δ
‑
mno2,目标催化剂α/a
‑
mno2‑
3和参比样品α
‑
mno2‑
3和a
‑
mno2的甲醛催化氧化性能图。
34.图5为本发明实施例1中所得目标催化剂α/a
‑
mno2‑
3的稳定性测试结果图。
35.图6为本发明实施例2中不同球磨时间所得系列催化剂α/a
‑
mno2‑
t的x射线衍射图;其中,t指球磨时间(h)。
36.图7为本发明实施例2中不同球磨时间所得系列催化剂α/a
‑
mno2‑
t的甲醛催化氧化性能图。
37.图8为本发明实施例3中不同球料比所得系列催化剂α/a
‑
mno2‑
r的x射线衍射图;其中,r指球料比。
38.图9为本发明实施例3中不同球料比所得系列催化剂α/a
‑
mno2‑
r的甲醛催化氧化性能图。
39.图10为本发明对比例1中不同晶型mno2经球磨处理后所得催化剂的x射线衍射图。
40.图11为本发明对比例1中不同晶型mno2经球磨处理后所得催化剂的甲醛催化氧化性能图。
具体实施方式
41.下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式和保护范围不限于此。
42.实施例1
43.(1)催化剂制备:
44.采用水热法制得前躯体δ
‑
mno2,具体步骤为:将15.0mmol kmno4、2.5mmol mnso4分散于120ml超纯水中,先后经超声、搅拌各1小时后置于容积为200ml聚四氟乙烯反应釜中,
经160℃恒温反应12小时后自然冷却至室温,所得沉淀物经充分清洗(分别用超纯水、无水乙醇清洗)、干燥后制得前躯体δ
‑
mno2。
45.α/a
‑
mno2‑
3异相结催化剂的制备:采用球磨法制得目标催化剂。具体为,在玛瑙罐中加入0.1g经水热所得δ
‑
mno2,球料比为100:1。使用行星式球磨机将前躯体δ
‑
mno2在600rpm的转速下,球磨3h,制得目标催化剂。
46.参比样品α
‑
mno2‑
3的制备:采用水热结合球磨的方法制得。其中水热法与前躯体样品δ
‑
mno2的制备方法类似,区别在于kmno4和mnso4摩尔比由6:1改为2.5:1,其它相同得到α
‑
mno2。随后将α
‑
mno2样品在与目标催化剂一致的球磨条件下处理,制得参比样品。
47.参比样品非晶相锰氧化物(记为:a
‑
mno2)的制备:在玛瑙罐中加入经水热所得δ
‑
mno2和0.01g石墨烯氧化物,其余球磨条件与目标催化剂一致,制得参比样品。
48.(2)催化剂的物相/结构/元素化学态表征:
49.本实施例所得催化剂α/a
‑
mno2‑
3的x射线衍射和选区电子衍射图分别如图1和图2中的b所示。结合xrd和选区电子衍射分析证明,δ
‑
mno2在机械力的作用下最终转变为α
‑
mno2,该相变过程经历非晶中间相,制得的催化剂α/a
‑
mno2‑
3含有非晶相锰氧化物和α
‑
mno2构成的相界面。高分辨透射电镜观察(见图2中的c)进一步证明非晶相/alpha相异相结的存在。透射电镜观察(见图2中的a)发现,研磨后的样品呈不规则颗粒状。
50.根据x射线光电子能谱分析(见图3a、图3b、图3c),所得目标催化剂α/a
‑
mno2‑
3的o1s谱表明该催化剂表面存在大量的氧空位;此外,mn 2p与3s谱中均证明存在相应低价态的mn
3
信号,进一步确证了催化剂表面存在氧空位。
51.(3)本实施例所得催化剂α/a
‑
mno2‑
3催化性能测试:
52.不同温度下目标催化剂、前躯体δ
‑
mno2及参比样品α
‑
mno2‑
3和a
‑
mno2‑
3的催化活性变化(见图4)表明,α/a
‑
mno2‑
3催化剂具有优异的催化反应活性,在80℃下即可将120ppm的甲醛完全催化氧化为co2和h2o,说明其具有优异的低温催化活性。其活性优于多数目前报道的锰基非贵金属催化剂。甲醛催化氧化反应条件:原料为120ppm甲醛与高纯空气混合气,气体体积空速为200l g
cat
‑1h
‑1。
53.图5给出了α/a
‑
mno2‑
3催化剂的稳定性测试结果,经过24小时等温(80℃)测试,催化剂活性未现衰退,说明催化剂具备优异的稳定性。甲醛催化氧化反应条件:原料为120ppm甲醛与高纯空气混合气,气体体积空速为300l g
cat
‑1h
‑1。
54.实施例2
55.(1)催化剂制备:
56.本实施例的合成方法中,仅将球磨时间改为0.5h、1h、3h、10h和20h,其余制备条件与实施例1一致。所得催化剂记为α/a
‑
mno2‑
t,其中t为不同球磨时间。
57.(2)催化剂的物相/结构表征:
58.本实施例所得催化剂α/a
‑
mno2‑
t的x射线衍射如图6所示。根据xrd分析,制得的催化剂α/a
‑
mno2‑
t随球磨时间延长,属于α
‑
mno2的衍射峰增强,意味着在球磨过程中δ
‑
mno2由向α
‑
mno2转变,经历非晶的中间相后,大部分转变为α
‑
mno2。其中催化剂α/a
‑
mno2‑
3含有大量非晶相锰氧化物和α
‑
mno2构成的相界面。
59.(3)本实施例所得催化剂α/a
‑
mno2‑
t催化性能测试:
60.不同温度下催化剂的催化性能变化(见图7)表明,α/a
‑
mno2‑
t催化剂随球磨时间
的延长,甲醛催化氧化的活性提高;当球磨时间超过3h,到达10~20h时,甲醛催化活性不再提高,且有略微下降。其中α/a
‑
mno2‑
3催化剂具有优异的催化反应活性,在80℃时即可将120ppm的甲醛完全催化氧化为co2和h2o,表明该催化剂具有较好的低温催化活性;此外,其表现出较高的催化反应速率(0.21μmol m
‑2min
‑1)其活性优于多数目前报道的锰基非贵金属催化剂。甲醛催化氧化反应条件:原料为120ppm甲醛与高纯空气混合气,气体体积空速为200l g
cat
‑1h
‑1。
61.实施例3
62.(1)催化剂制备:
63.本实施例的合成方法中,仅将球料比改为10:1、100:1和200:1,其余制备条件与实施例1一致。所得催化剂记为α/a
‑
mno2‑
r,其中r为不同球料比。
64.(2)催化剂的物相/结构表征:
65.本实施例所得催化剂α/a
‑
mno2‑
r的x射线衍射如图8所示。根据xrd分析,不同球料比皆能实现δ
‑
mno2到α
‑
mno2相变,其中制得的催化剂α/a
‑
mno2‑
r随球料比增加,属于α
‑
mno2的衍射峰增强,较大的球料比意味着较高的机械强度,因而加速球磨中的相变过程。
66.(3)本实施例所得催化剂α/a
‑
mno2‑
r催化性能测试:
67.不同温度下催化剂的催化性能变化(图9)表明,α/a
‑
mno2‑
r催化剂催化氧化甲醛的活性与球料比有关,当球料比为100:1时最佳,而过低或过高的球料比皆不利于提高甲醛催化的活性。其中α/a
‑
mno2‑
100催化剂在80℃时即可将120ppm的甲醛完全催化氧化为co2和h2o,表明该催化剂具有较好的低温催化活性;其活性优于多数目前报道的锰基非贵金属催化剂。甲醛催化氧化反应条件:原料为120ppm甲醛与高纯空气混合气,气体体积空速为200l g
cat
‑1h
‑1。
68.对比例1
69.(1)催化剂制备:
70.目标催化剂δ
‑
mno2‑
bm(bm为球磨法)的制备:以δ
‑
mno2为前躯体球磨,仅将球磨时间改为1h,其余制备条件与实施例1一致。所得目标催化剂记为δ
‑
mno2‑
bm。
71.参比样品α
‑
mno2‑
bm的制备:以α
‑
mno2为前躯体球磨,仅将球磨时间改为1h,其余制备条件与实施例1一致。所得目标催化剂记为α
‑
mno2‑
bm。
72.参比样品β
‑
mno2‑
bm的制备:取0.1g商用β
‑
mno2球磨,球磨时间为1h,其余球磨条件与实例1一致。所得目标催化剂记为β
‑
mno2‑
bm。
73.(2)催化剂的物相/结构表征:
74.本实施例所得催化剂的x射线衍射如图10所示。根据xrd分析,δ
‑
mno2经球磨后发生明显相变行为,而α
‑
mno2和β
‑
mno2经球磨处理后无相变现象。
75.(3)本实施例所得催化剂催化性能测试:
76.不同温度下催化剂的催化性能变化(图11)表明,甲醛催化氧化的活性与前躯体的晶型有关。δ
‑
mno2‑
bm催化剂较参比样品而言表现出优异的甲醛催化的活性,在100℃时即可将120ppm的甲醛完全催化氧化为co2和h2o,表明该催化剂具有较好的低温催化活性;其活性优于多数目前报道的锰基非贵金属催化剂。甲醛催化氧化反应条件:原料为120ppm甲醛与高纯空气混合气,气体体积空速为200l g
cat
‑1h
‑1。
77.上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的
限制,其它的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。