1.本公开涉及用于图像形成方法例如电子照相法的调色剂。
背景技术:
2.近年来,对电子照相图像形成装置提出了对于高速化、长寿命化、节能化和小型化更高的要求,并且,响应于这些要求,还要求调色剂的各种特性的进一步改善。在高速化和长寿命化方面,显影装置内的调色剂倾向于经受增加的对例如热和冲击等应力的暴露。结果,为了即使在输出多张图像期间无论使用环境如何均保持良好的图像品质,需要可以将带电性能和耐久性保持在高水平的高耐久性调色剂。
3.另一方面,在节能化方面,需要显示优异的低温定影性的调色剂,尽管这通常涉及与高耐久性的折衷。结果,对能够以高水平显示高耐久性和低温定影性二者的调色剂存在更大需求。
4.用于解决该问题的一种手段为用树脂覆盖调色剂颗粒的表面的方法。
5.作为用硅化合物覆盖调色剂颗粒表面的方法,日本专利申请特开no.h03
‑
089361记载了将硅烷偶联剂添加至反应体系中的聚合调色剂的生产方法。
6.日本专利申请特开no.h09
‑
179341记载了在表面上具有来自自由基聚合性有机硅烷化合物的反应产物的涂膜的聚合调色剂。
技术实现要素:
7.根据由本发明人进行的研究的结果,在日本专利申请特开no.h03
‑
089361中记载的调色剂的情况下,调色剂表面上的硅烷化合物的沉积量不足,并且对于高速化和长寿命化所需的低温定影性和高耐久性的共存存在改进空间。在日本专利申请特开no.h09
‑
179341中记载的调色剂的情况下,有机官能团具有高的极性并且调色剂颗粒表面上的硅烷化合物的沉积量不足,并且硅烷化合物的水解和缩聚不足,此外,交联度弱。结果,这对于与高速化和长寿命化相关的低温定影性和高耐久性是不充分的,并且存在改进空间。
8.本公开提供即使在图像形成装置的高速化和长寿命化的情况下也显示低温定影性和高耐久性的调色剂。
9.作为解决上述问题的深入研究的结果,本发明人发现可以通过以下描述的调色剂来解决该问题。
10.本公开涉及一种调色剂,其包含调色剂颗粒,所述调色剂颗粒包括
11.含有粘结剂树脂的核颗粒,和
12.含有无机细颗粒和有机硅聚合物的表层,其中
13.有机硅聚合物具有由下式(t3)给出的结构:
14.r
‑
si(o
1/2
)3ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(t3)
15.其中,r表示具有1至6个碳的烷基或苯基;
16.在调色剂颗粒的四氢呋喃不溶性物质的
29
si
‑
nmr测量中,归属于由式(t3)给出的
结构的峰面积相对于有机硅聚合物的总峰面积的比例为至少5.0%;和
17.在使用透射型电子显微镜的对调色剂颗粒的截面的观察中,
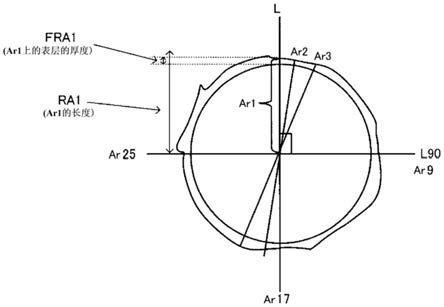
18.长轴l为穿过调色剂颗粒的几何中心并且具有调色剂颗粒的截面中的最长直径的弦,
19.线段a为将长轴l在其中点处分割时的线段之一,
20.arn(n=1至32)分别为以线段a作为基准每次偏移11.25
°
而从长轴l的中点至调色剂颗粒的表面为止绘制的32条线段,
21.ran(n=1至32)为各线段的长度,和
22.fran(n=1至32)为arn(n=1至32)上的表层的厚度,
23.在按照下式(1)来定义的dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒的截面中,
24.(i)表层的平均厚度dav.为5.0至100.0nm,
25.(ii)fran不超过5.0nm的arn线段的比例不超过20.0%,
26.(iii)表层中与核颗粒接触的无机细颗粒的数量为每1个调色剂颗粒16个至30个,和
27.(iv)在100个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒中,包含至少一个存在于核颗粒中并且不与表层接触的无机细颗粒的调色剂颗粒的比例不超过10%:
28.dtem=(ra1 ra2 ra3 ra4 ra5 ra6 ra7 ra8 ra9 ra10 ra11 ra12 ra13 ra14 ra15 ra16 ra17 ra18 ra19 ra20 ra21 ra22 ra23 ra24 ra25 ra26 ra27 ra28 ra29 ra30 ra31 ra32)/16
ꢀꢀꢀꢀ
(1)。
29.本公开由此可以提供即使在图像形成装置的高速化和长寿命化的情况下也显示稳定的带电性能和高耐久性的调色剂。
30.参考附图,本发明的进一步的特征将从以下示例性实施方案的描述变得显而易见。
附图说明
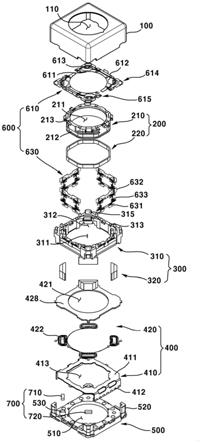
31.图1为图像形成装置的示意性结构图的实例;
32.图2为如使用透射型电子显微镜观察到的调色剂颗粒截面的图像的实例;和
33.图3为调色剂颗粒的thf不溶性物质的
29
si
‑
nmr测量的实例。
具体实施方式
34.以下详细地描述了本公开。在图中使用的附图标记定义如下。
35.11感光构件、12显影辊、13调色剂供给辊、14调色剂、15调节刮板、16显影设备、17激光、18充电设备、19清洁设备、20清洁用充电设备、21搅拌叶片、22驱动辊、23转印辊、24偏置电源、25张力辊、26转印输送带、27从动辊、28纸、29供纸辊、30吸附辊、31定影设备
36.本公开涉及一种调色剂,其包含调色剂颗粒,所述调色剂颗粒包括
37.含有粘结剂树脂的核颗粒,和
38.含有无机细颗粒和有机硅聚合物的表层,其中
39.有机硅聚合物具有由下式(t3)给出的结构:
40.r
‑
si(o
1/2
)3ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(t3)
41.其中,r表示具有1至6个碳的烷基或苯基;
42.在调色剂颗粒的四氢呋喃不溶性物质的
29
si
‑
nmr测量中,归属于由式(t3)给出的结构的峰面积相对于有机硅聚合物的总峰面积的比例为至少5.0%;和
43.在使用透射型电子显微镜的对调色剂颗粒的截面的观察中,
44.长轴l为穿过调色剂颗粒的几何中心并且具有调色剂颗粒的截面中的最长直径的弦,
45.线段a为将长轴l在其中点处分割时的线段之一,
46.arn(n=1至32)分别为以线段a作为基准每次偏移11.25
°
而从长轴l的中点至调色剂颗粒的表面为止绘制的32条线段,
47.ran(n=1至32)为各线段的长度,和
48.fran(n=1至32)为arn(n=1至32)上的表层的厚度,
49.在按照下式(1)来定义的dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒的截面中,
50.(i)表层的平均厚度dav.为5.0至100.0nm,
51.(ii)fran不超过5.0nm的arn线段的比例不超过20.0%,
52.(iii)表层中与核颗粒接触的无机细颗粒的数量为每1个调色剂颗粒16个至30个,和
53.(iv)在100个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒中,包含至少一个存在于核颗粒中并且不与表层接触的无机细颗粒的调色剂颗粒的比例不超过10%:
54.dtem=(ra1 ra2 ra3 ra4 ra5 ra6 ra7 ra8 ra9 ra10 ra11 ra12 ra13 ra14 ra15 ra16 ra17 ra18 ra19 ra20 ra21 ra22 ra23 ra24 ra25 ra26 ra27 ra28 ra29 ra30 ra31 ra32)/16
ꢀꢀꢀꢀ
(1)。
55.本发明人认为以下是当满足指定条件时获得本公开的效果的原因。
56.调色剂颗粒具有包含无机细颗粒和有机硅聚合物的表层,并且有机硅聚合物具有由r
‑
sio
3/2
(式(t3))给出的结构。
57.式中的r表示具有1个至6个碳(优选1个至4个碳并且更优选1个或2个碳)的烷基、或苯基。
58.在由式(t3)给出的结构中,硅原子的四个化合价中的一个与由r表示的有机基团键合并且其余三个与氧原子键合。氧原子处于它们的两个化合价均与硅原子键合的状态,即,硅氧烷键(si
‑
o
‑
si)。当考虑有机硅聚合物形式的硅原子和氧原子时,由于对于两个硅原子存在三个氧原子,因此用
‑
sio
3/2
来表示。即,由式(t3)表示的结构为如由下式给出的结构。
[0059][0060]
该
‑
sio
3/2
结构与由大量硅氧烷结构构成的二氧化硅(sio2)类似,并且,由于其具有与二氧化硅类似的硬度,本发明人认为可以通过将
‑
sio
3/2
结构引入至调色剂颗粒的表层中的有机硅聚合物中来实现高耐久性。
[0061]
此外,在调色剂颗粒的四氢呋喃不溶性物质(以下也称为thf不溶性物质)的
29
si
‑
nmr测量中,归属于由式(t3)给出的结构的峰面积相对于有机硅聚合物的总峰面积的比例为至少5.0%。虽然以下给出了测量方法的细节,但是这近似意味着,在调色剂颗粒的表层中包含的有机硅聚合物中,参与由r
‑
sio
3/2
给出的结构的硅原子的比例为有机硅聚合物中的全部硅原子的至少5.0%。
[0062]
如上所述,由r
‑
sio
3/2
给出的结构的含义为,在硅原子的四个化合价中,三个与氧原子键合并且这些氧原子与不同的硅原子键合。当这些氧原子中的一个构成硅烷醇基团时,则该结构由r
‑
sio
2/2
‑
oh表示。该结构类似于由二甲基硅酮表示的二取代硅酮树脂。
[0063]
因此,认为随着存在更多的由r
‑
sio
3/2
给出的结构,调色剂颗粒的表层如上所述开始显示如二氧化硅那样的硬度性质并且可以显示高耐久性。另一方面,当存在较少的r
‑
sio
3/2
结构时,例如,随着由r
‑
sio
2/2
‑
oh表示的结构增加,有机特性变得占优势并且耐久性下降,而随着由sio2给出的结构变得更多,如二氧化硅那样的硬度性质变得占优势并且定影性能下降。
[0064]
结果,有机硅聚合物必须具有至少5.0%的由r
‑
sio
3/2
给出的结构。即,归属于由式(t3)给出的结构的峰面积相对于有机硅聚合物的总峰面积的比例必须为至少5.0%。该峰面积的比例可以为例如不超过85.0%.
[0065]
从高耐久性的观点来看,归属于具有式(t3)的结构的峰面积相对于有机硅聚合物的总峰面积的比例优选为20.0%至85.0%,更优选40.0%至80.0%,并且甚至更优选40.0%至67.5%。可以使用在形成具有式(t3)的结构期间的反应温度和使用反应期间的ph来控制由式(t3)给出的结构的峰面积的比例。
[0066]
此外,在使用透射型电子显微镜(以下也称为tem)的对调色剂颗粒截面的观察中,
[0067]
长轴l为穿过调色剂颗粒的几何中心并且具有调色剂颗粒的截面中的最长直径的弦,
[0068]
线段a为将长轴l在其中点处分割时的线段之一,
[0069]
arn(n=1至32)分别为以线段a作为基准每次偏移11.25
°
而从长轴l的中点至调色剂颗粒的表面为止绘制的32条线段,
[0070]
ran(n=1至32)为各线段的长度,和
[0071]
fran(n=1至32)为arn(n=1至32)上的表层的厚度,
[0072]
可以通过使按照以下式(1)来定义的dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒的截面中表层的平均厚度dav.为5.0nm至100.0nm来使高水平的耐久性与高水平的定影性能共存。
[0073]
dtem=(ra1 ra2 ra3 ra4 ra5 ra6 ra7 ra8 ra9 ra10 ra11 ra12 ra13 ra14 ra15 ra16 ra17 ra18 ra19 ra20 ra21 ra22 ra23 ra24 ra25 ra26 ra27 ra28 ra29 ra30 ra31 ra32)/16
ꢀꢀꢀꢀꢀꢀ
(1)
[0074]
当该dav.低于5.0nm时耐久性下降,而当该dav.超过100.0nm时定影性能下降。该dav.优选为10.0nm至70.0nm并且更优选为10.0nm至50.0nm。可以通过例如有机硅聚合物的含量、有机硅聚合物中亲水性基团与疏水性基团的比例、以及水解、加成聚合和缩聚期间的反应温度、反应时间、反应介质和ph来控制有机硅聚合物的表层的平均厚度dav.。
[0075]
此外,能够耐受显影装置内的应力的高耐久性调色剂可以通过使dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒的截面中fran不超过5.0nm的arn线段的比例不超过20.0%来获得。当fran不超过5.0nm的arn线段的比例超过20.0%时,调色剂耐久性下降,在显影辊的表面上发生成膜,并且发生浓淡不均匀。fran不超过5.0nm的arn线段的比例优选为不超过10.0%并且更优选不超过5.0%。fran不超过5.0nm的arn线段的比例优选尽可能低,并且为例如至少0.0%。可以使用这些数值范围的任意组合。
[0076]
可以通过例如有机硅聚合物的含量、有机硅聚合物中亲水性基团与疏水性基团的比例、以及在水解、加成聚合和缩聚期间的反应温度、反应时间、反应介质和ph来控制fran不超过5.0nm的arn线段的比例。
[0077]
然而,具有如二氧化硅那样的硬度性质的具有r
‑
sio
3/2
结构的有机硅聚合物在调色剂颗粒表层中的存在引起关于对定影性能的影响的担忧。作为深入研究的结果,本发明人发现,还可以通过使表层中与核颗粒接触的无机细颗粒的数量为每1个调色剂颗粒16个至30个来实现与定影性能的共存。
[0078]
无机细颗粒通常表现出硬质,并且,认为结果是当调色剂颗粒中包含无机细颗粒时,无机细颗粒会使定影性能降低。然而,通过满足指定条件,认为在定影期间,无机细颗粒破坏表层并且有助于调色剂的熔融,并且结果是为优异的定影性能提供支持。
[0079]
当无机细颗粒的数量小于16个时,无法充分地破坏表层,从而使定影性能降低。当无机细颗粒的数量超过30个时,调色剂的带电性能最终降低并且因此发生起雾。无机细颗粒的数量优选为19个至29个并且更优选为20个至24个。
[0080]
表层中与核颗粒接触的无机细颗粒的数量可以使用例如在造粒步骤期间补充添加的用作分散稳定剂的无机细颗粒的量来控制。
[0081]
此外,在100个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒中,具有至少一个存在于核颗粒中并且不与表层接触的无机细颗粒的调色剂颗粒的比例必须不超过10%。当具有存在于核颗粒中并且不与表层接触的无机细颗粒的调色剂颗粒的比例超过10%时,这引起如上所述的定影性能的降低。此类调色剂颗粒的比例优选为不超过5%并且更优选不超过2%。此类调色剂颗粒的比例优选尽可能低,并且例如为至少0%。可以使用这些数值范围的任意组合。
[0082]
具有至少一个存在于核颗粒中并且不与表层接触的无机细颗粒的调色剂颗粒的比例可以通过例如在造粒步骤期间补充添加的用作分散稳定剂的无机细颗粒的量来控制。
[0083]
作为无机细颗粒,可以使用已知的无机细颗粒而没有特别限制,但是无机细颗粒优选包含选自由钙元素和镁元素组成的组中的至少一者,包含钙元素是更优选的。
[0084]
无机细颗粒中选自由钙元素和镁元素组成的组中的至少一者(优选钙元素)的含量优选为50质量%至100质量%并且更优选85质量%至100质量%。
[0085]
使用dm作为无机细颗粒的一次粒径的数均粒径,dm优选为50.0nm至800.0nm。通过使dm为至少50.0nm,促进在定影期间表层的破坏并且确立在定影性能方面进一步改善的倾向。通过使dm不超过800.0nm,可以抑制对调色剂带电性能的影响并且确立进一步抑制起雾的发生的倾向。dm更优选为90.0nm至200.0nm。可以通过例如在制备补充添加的用作分散稳定剂的无机细颗粒期间的温度和搅拌器旋转速度来控制该dm。
[0086]
在调色剂颗粒截面的tem观察中,在100个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒中,在表层的夹在ar1和ar5之间的区域、表层的夹在ar5和ar9之间的区域、表层的夹在ar9和ar13之间的区域、表层的夹在ar13和ar17之间的区域、表层的夹在ar17和ar21之间的区域、表层的夹在ar21和ar25之间的区域、表层的夹在ar25和ar29之间的区域、和表层的夹在ar29和ar1之间的区域(以下,这些区域也统一简称为“表层的八等分区域”)中的每一者中存在至少一个无机细颗粒的调色剂颗粒的比例为至少90%。
[0087]
结果,在定影期间可以使调色剂颗粒的表层被完全破坏并且可以建立甚至更好的定影性能。这些调色剂颗粒的比例更优选为至少95%。这些调色剂颗粒的比例优选尽可能高,例如,不超过100%。可以使用这些数值范围的任意组合。
[0088]
可以通过例如在造粒步骤期间补充添加的用作分散稳定剂的无机细颗粒的量和在造粒步骤期间的搅拌旋转速度来控制这些调色剂颗粒的比例。
[0089]
在调色剂颗粒截面的tem观察中,在100个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒中,表层中接触核颗粒的无机细颗粒的数量为16个至30个的调色剂颗粒的比例优选为至少90%。这用于提供在定影期间有机硅聚合物表层的破坏良好地进行的大量调色剂,并且结果是提供甚至更好的定影。至少95%是更优选的。该调色剂颗粒比例优选尽可能高,并且例如为不超过100%。可以使用这些数值范围的任意组合。
[0090]
表层的平均厚度dav.和无机细颗粒的一次粒径dm优选满足dav./dm<1.00。通过满足dav./dm<1.00,使无机细颗粒的粒径相对于表层的平均厚度足够大,结果,促进借助无机细颗粒来破坏表层并且提供甚至更好的定影性能。dav./dm≤0.80是更优选的。
[0091]
以下详细地描述本公开的实施方案。
[0092]
以下化合物为用于生产有机硅聚合物的有机硅化合物的具体实例:甲基三甲氧基硅烷、甲基三乙氧基硅烷、甲基三氯硅烷、乙基三甲氧基硅烷、乙基三乙氧基硅烷、乙基三氯硅烷、乙基三乙酰氧基硅烷、丙基三甲氧基硅烷、丙基三乙氧基硅烷、丙基三氯硅烷、丁基三甲氧基硅烷、丁基三乙氧基硅烷、丁基三氯硅烷、丁基甲氧基二氯硅烷、丁基乙氧基二氯硅烷、己基三甲氧基硅烷、己基三乙氧基硅烷、苯基三甲氧基硅烷、和苯基三乙氧基硅烷。这些有机硅化合物中的单独一种可以单独使用或者其中至少两种可以组合使用。
[0093]
已知在溶胶
‑
凝胶反应中,生成的硅氧烷键的键合状态通常随反应介质的酸度的变化而变化。
[0094]
具体地,当介质为酸性时,氢离子亲电地加成至一个反应性基团(例如,烷氧基)中的氧。然后,水分子中的氧原子与硅原子配位并且通过取代反应发生向氢硅烷基的转化。假
定存在足够的水,由于反应性基团(例如,烷氧基)的一个氧原子被一个h
攻击,因此,当反应介质中的h
含量低时,产生羟基的取代反应将变慢。因此,缩聚反应在硅烷中键合的所有反应性基团水解之前发生,从而相对容易地生成一维线状高分子或二维高分子。
[0095]
另一方面,当介质为碱性时,氢氧根离子经由五配位中间体加成至硅。因此,所有反应性基团(例如,烷氧基)容易被消除并且容易被硅烷醇基团取代。特别地,当使用在同一硅烷中具有至少三个反应性基团的硅化合物时,水解和缩聚在三维上进行并且形成具有丰富的三维交联键的有机硅聚合物。此外,反应也在短时间内完成。
[0096]
因此,用于形成有机硅聚合物的溶胶
‑
凝胶反应优选在碱性条件下进行,并且,具体地,当在水系介质中进行生产时,优选反应在至少8.0的ph和至少90℃的反应温度下进行,反应时间为至少5小时。这样做为形成具有较高的强度和优异的耐久性的有机硅聚合物提供支持。
[0097]
当用于上述悬浮聚合的介质为水系介质时,可以使用以下无机细颗粒作为用于聚合性单体组合物的颗粒的分散稳定剂:例如,磷酸三钙、磷酸镁、碳酸钙、碳酸镁、氢氧化钙、氢氧化镁、偏硅酸钙、和硫酸钙。
[0098]
可以将任何生产方法用于调色剂颗粒生产的生产方法,但是悬浮聚合法是优选的。
[0099]
以下以借助悬浮聚合法的调色剂颗粒生产方法为实例,并且,虽然详细地描述了调色剂颗粒生产方法,但是调色剂颗粒生产方法绝不限于以下方法或者受以下方法限制。
[0100]
悬浮聚合法为如下生产方法:将包含聚合性单体和任选的着色剂的聚合性单体组合物添加至水系介质中;将水系介质中的聚合性单体组合物造粒以形成聚合性单体组合物的颗粒;并且使聚合性单体组合物的颗粒中包含的聚合性单体聚合以获得调色剂颗粒。
[0101]
以下描述使用悬浮聚合法的调色剂颗粒生产方法中的每个步骤。
[0102]
(聚合性单体组合物的准备)
[0103]
制备包含聚合性单体和有机硅化合物以及任选的着色剂的聚合性单体组合物。当聚合性单体组合物包含着色剂时,可以将着色剂在使用例如介质搅拌磨机(stirred media mill)在聚合性单体中初步分散之后与其它组合物混合,或者可以在混合该其它组合物的同时或者在混合该其它组合物之后进行分散。
[0104]
(造粒步骤)
[0105]
将聚合性单体组合物投入至包含如上所述用作分散稳定剂的无机细颗粒的水系介质中,并且通过分散将水系介质中的聚合性单体组合物造粒成液滴以获得聚合性单体组合物的液滴。
[0106]
在造粒步骤期间,无机细颗粒附着至聚合性单体组合物液滴的表面。当聚合性单体组合物包含极性树脂时,该极性树脂被附着至液滴表面的无机细颗粒吸引至界面。虽然极性树脂由于其极性而天然地倾向于存在于聚合性单体组合物的表面,但是该吸引至界面的效果进一步促进极性树脂存在于表面。
[0107]
可以例如使用安装有产生高剪切力的搅拌机的立式搅拌槽来进行造粒步骤。可以使用例如,市售的高剪切搅拌机,例如,ultra
‑
turrax(ika)、t.k.均质混合器(tokushu kika kogyo co.,ltd.)、t.k.filmics(tokushu kika kogyo co.,ltd.)、clearmix(m technique co.,ltd.)、cavimix(pacific machinery&engineering co.,ltd.)等作为产生
高剪切力的搅拌机。还可用作搅拌机的是具有循环机构并且可以在循环机构内产生串列式(inline)高剪切力的分散机,其中循环机构从搅拌槽的下部除去立式搅拌槽内的一部分处理液并且使其返回至搅拌槽。可以使用例如colloid mill(ika)、cavitron(pacific machinery&engineering co.,ltd.)、w motion(m technique co.,ltd.)等市售分散机作为串列式分散机。在造粒步骤期间,还可以补充添加用作分散稳定剂的无机细颗粒。这起到促进表层中与核颗粒接触的无机细颗粒的存在的作用。
[0108]
(聚合步骤)
[0109]
将由此获得的聚合性单体组合物的分散液引入至聚合步骤中以获得调色剂颗粒分散液。可以在聚合步骤中使用普通的温度可控的搅拌槽。
[0110]
聚合温度为至少40℃并且优选为50℃至90℃。聚合温度可以自始至终保持恒定,或者可以在聚合步骤的后半段升高,目的是获得期望的分子量分布。用于搅拌的搅拌叶片可以为支持保持槽内均一的温度、可以使调色剂用原料分散液悬浮并且这样做不会使该原料分散液滞留的任何搅拌叶片。搅拌叶片或搅拌装置可以示例为普通的搅拌叶片,例如桨叶片(paddle blade)、倾斜的桨叶片(pitched paddle blade)、三叶后退螺旋桨(swept three
‑
blade propeller)、螺旋桨叶片(propeller blade)、圆盘涡轮叶片(disk turbine blade)、螺旋带叶片(helical ribbon blade)、锚叶片(anchor blade)等,并且可以示例为“fullzone”(shinko pantec co.,ltd.)、“twinstar”(shinko pantec co.,ltd.)、“maxblend”(sumitomo heavy industries,ltd.)、“super mix”(satake chemical equipment mfg.,ltd.)和“hi
‑
f混合机”(soken chemical&engineering co.,ltd.)。
[0111]
此外,通过将聚合步骤中的温度和ph控制至上述范围内,可以使聚合性单体组合物中的有机硅化合物进行溶胶
‑
凝胶反应并且可以形成包含无机细颗粒和有机硅聚合物的表层。
[0112]
(蒸馏步骤)
[0113]
为了根据需要除去挥发性杂质,如,例如,未反应的聚合性单体、副产物等,在完成聚合之后可以在蒸馏步骤中将一部分水系介质蒸馏掉。蒸馏步骤可以在常压下或在减压下进行。
[0114]
(清洗步骤、固液分离步骤和干燥步骤)
[0115]
还可以用酸或碱来处理聚合物颗粒分散液,目的是除去附着至聚合物颗粒表面的过量的分散稳定剂。此后,使用普通的固液分离法将聚合物颗粒从液相中分离,并且用新添加的水洗涤聚合物颗粒从而除去酸或碱和溶解于其中的过量的分散稳定剂组分。通过重复该清洗步骤数次来进行充分洗涤并且再次进行固液分离以获得调色剂颗粒。根据需要,可以使用已知的干燥手段来干燥获得的调色剂颗粒。
[0116]
(分级步骤)
[0117]
当由此获得的调色剂颗粒要求更尖锐的粒度时,还可以通过使用例如风力分级机进行分级来将期望的粒度分布以外的颗粒分级并且除去。
[0118]
以下乙烯基系聚合性单体为上述聚合性单体的优选实例:苯乙烯;苯乙烯衍生物,例如,α
‑
甲基苯乙烯、β
‑
甲基苯乙烯、邻甲基苯乙烯、间甲基苯乙烯、对甲基苯乙烯、2,4
‑
二甲基苯乙烯、对正丁基苯乙烯、对叔丁基苯乙烯、对正己基苯乙烯、对正辛基苯乙烯、对正壬基苯乙烯、对正癸基苯乙烯、对正十二烷基苯乙烯、对甲氧基苯乙烯、和对苯基苯乙烯;丙烯
yellow s)、汉萨黄g(hansa yellow g)、汉萨黄10g、联苯胺黄g、联苯胺黄gr、喹啉黄色淀、例如永固黄ncg和酒石黄色淀等缩合偶氮化合物、异吲哚啉酮化合物、蒽醌化合物、偶氮金属络合物、次甲基化合物、和烯丙基酰胺化合物作为黄色颜料。具体实例如下:c.i.颜料黄12、13、14、15、17、62、74、83、93、94、95、109、110、111、128、129、147、155、168和180。
[0128]
橙色颜料可以示例为以下物质:永固橙gtr、吡唑啉酮橙、硫化橙(vulcan orange)、联苯胺橙g、阴丹士林亮橙rk、和阴丹士林亮橙gk。
[0129]
例如,红色铁氧化物;缩合偶氮化合物,例如永固红4r、立索尔红、吡唑啉酮红、表红钙盐(watching red calcium salt)、色淀红c、色淀红d、亮胭脂红6b、亮胭脂红3b、曙红色淀、罗丹明色淀b、和茜素色淀;二酮吡咯并吡咯化合物;蒽醌;喹吖啶酮化合物;碱性染料色淀化合物;萘酚化合物;苯并咪唑酮化合物;硫靛化合物;和苝化合物为红色颜料的实例。
[0130]
蓝色颜料可以示例为碱性蓝色淀;维多利亚蓝色淀(victoria blue lake);铜酞菁化合物及其衍生物,例如,酞菁蓝、无金属酞菁蓝、部分氯化的酞菁蓝、坚牢天蓝(fast sky blue)、和阴丹士林bg;蒽醌化合物;和碱性染料色淀化合物。具体实例如下:c.i.颜料蓝1、7、15、15:1、15:2、15:3、15:4、60、62和66。
[0131]
紫色颜料可以示例为固紫b(fast violet b)和甲基紫色淀。
[0132]
绿色颜料可以示例为颜料绿b、孔雀石绿色淀和最终黄绿g(final yellow green g)。
[0133]
白色颜料可以示例为锌白、氧化钛、锑白和硫化锌。
[0134]
黑色颜料可以示例为炭黑、苯胺黑、非磁性铁氧体和磁铁矿、以及通过使用上述黄色系着色剂、红色系着色剂和蓝色系着色剂调色得到黑色来提供的黑色颜料。
[0135]
这些着色剂中的单独一种可以单独使用或者可以使用其中至少两种的组合。这些着色剂还可以以固溶体形式使用。着色剂含量相对于100质量份粘结剂树脂或聚合性单体优选为3.0质量份至15.0质量份。
[0136]
可以在调色剂中使用具有在调色剂生产期间具有特定pka的离子性官能团的树脂以外的电荷控制剂。可以使用已知的电荷控制剂作为该电荷控制剂。该电荷控制剂的添加量相对于100质量份粘结剂树脂或聚合性单体优选为0.01质量份至10.0质量份。
[0137]
可以原样使用调色剂颗粒作为调色剂,或者还可以通过向调色剂颗粒中任选外部添加各种有机细粉或无机细粉中的任意者来提供调色剂。考虑到向调色剂颗粒中添加时的耐久性,不超过调色剂颗粒的重均粒径的十分之一的粒径对于该有机或无机细粉是优选的。例如,将以下物质用于有机细粉或无机细粉。
[0138]
(1)流动性赋予剂:二氧化硅、氧化铝、氧化钛、炭黑、和氟化碳。
[0139]
(2)研磨剂:金属氧化物(例如,钛酸锶、氧化铈、氧化铝、氧化镁、氧化铬)、氮化物(例如,氮化硅)、碳化物(例如,碳化硅)、金属盐(例如,硫酸钙、硫酸钡、碳酸钙)。
[0140]
(3)润滑剂:氟系树脂粉末(例如,偏二氟乙烯、聚四氟乙烯)、脂肪酸的金属盐(例如,硬脂酸锌、硬脂酸钙)。
[0141]
(4)电荷控制性颗粒:金属氧化物(例如,氧化锡、氧化钛、氧化锌、二氧化硅、氧化铝)、炭黑。
[0142]
还可以使用有机细粉或无机细粉来处理调色剂颗粒的表面,从而改善调色剂流动性并且提供更均一的调色剂颗粒带电。用于对有机细粉或无机细粉进行疏水化处理的处理
剂可以示例为未改性的硅酮清漆、各种改性的硅酮清漆、未改性的硅油、各种改性的硅油、硅烷化合物、硅烷偶联剂、除了前述物质以外的有机硅化合物、和有机钛化合物。这些处理剂中的单独一种可以单独使用或者其中至少两种可以组合使用。
[0143]
以下描述用于测量本公开涉及的各物性值的方法。
[0144]
<用于分离nmr测量用调色剂颗粒中的thf不溶性物质的方法>
[0145]
如下来分离调色剂颗粒中的四氢呋喃(thf)不溶性物质。
[0146]
将10.0g调色剂颗粒称出并且置于抽提套管(no.86r,toyo roshi kaisha,ltd.)中,并且将其置于索氏提取器中。使用200ml thf作为溶剂进行提取20小时,并且将抽提套管中的过滤残余物在40℃下真空干燥数小时,以得到nmr测量用调色剂颗粒中的thf不溶性物质。当调色剂颗粒中包含磁性体时,例如在提取时可以使用磁石进行初步分离。
[0147]
当调色剂颗粒表面已经用例如外部添加剂进行处理时,通过使用以下方法除去外部添加剂来获得调色剂颗粒。
[0148]
通过将160g蔗糖(kishida chemical co.,ltd.)添加至100ml去离子水中并且在水浴上加热的同时使其溶解来制备蔗糖浓稠液。将31g该蔗糖浓稠液和6ml contaminon n(包含非离子表面活性剂、阴离子表面活性剂和有机助洗剂的ph 7的精密测量仪器清洗用中性清洁剂的10质量%水溶液,wako pure chemical industries,ltd.)引入至离心分离管中以制备分散液。将1.0g调色剂添加至该分散液中,并且使用例如刮刀将调色剂的团块打碎。
[0149]
将离心分离管用摇床以350每分钟冲程数(stroke per minute)(spm)振摇20分钟。在振摇后,将溶液转移至水平转子装置用玻璃管(50ml),并且使用3,500rpm和30分钟的条件用离心分离机进行分离。该过程将从调色剂颗粒脱离的外部添加剂分离。调色剂与水溶液的充分分离通过目视检查来确认,并且使用例如刮刀来回收分离至最上层中的调色剂。将回收的调色剂在减压过滤器上过滤然后在干燥机中干燥至少一小时以得到调色剂颗粒。
[0150]
多次进行该过程以确保所需量。
[0151]
(用于确认由式(t3)给出的结构的方法)
[0152]
使用以下方法来确认调色剂颗粒中包含的有机硅聚合物中的由式(t3)表示的结构。
[0153]
通过
13
c
‑
nmr和
29
si
‑
nmr来确认式(t3)中由r表示的烷基或苯基的有无。通过1h
‑
nmr、
13
c
‑
nmr和
29
si
‑
nmr来确认由式(t3)给出的结构的细节。以下给出所使用的仪器和测量条件。
[0154]
(1h
‑
nmr测量条件)
[0155]
仪器:来自bruker的avance iii 500
[0156]
探针:4mm mas bb/1h
[0157]
测量温度:室温
[0158]
样品旋转速率:6khz
[0159]
样品:将150mg测量样品(上述nmr测量用调色剂颗粒中的thf不溶性物质)引入至直径为4mm的样品管中。
[0160]
使用该方法来确认式(t3)中由r表示的烷基或苯基的有无。当可以确认到信号时,
将具有式(t3)的结构记为“有”。
[0161]
(
13
c
‑
nmr(固体状态)的测量条件)
[0162]
测量核频率:125.77mhz
[0163]
基准物质:甘氨酸(外部标准:176.03ppm)
[0164]
观察宽度:37.88khz
[0165]
测量方法:cp/mas
[0166]
接触时间:1.75msec
[0167]
重复时间:4秒
[0168]
扫描次数:2048次
[0169]
lb值:50hz
[0170]
(
29
si
‑
nmr(固体状态)的测量条件)
[0171]
仪器:来自bruker的avance iii 500
[0172]
探针:4mm mas bb/1h
[0173]
测量温度:室温
[0174]
样品旋转速率:6khz
[0175]
样品:将150mg测量样品(nmr测量用调色剂颗粒中的thf不溶性物质)引入至直径为4mm的样品管中。
[0176]
测量核频率:99.36mhz
[0177]
基准物质:dss(外部标准:1.534ppm)
[0178]
观察宽度:29.76khz
[0179]
测量方法:dd/mas、cp/mas
[0180]
29
si 90
°
[0181]
脉冲宽度:4.00μsec@
‑
1db
[0182]
接触时间:从1.75msec至10msec
[0183]
重复时间:30秒(dd/mas)、10秒(cp/mas)
[0184]
扫描次数:2048次
[0185]
lb值:50hz
[0186]
[用于测量归属于由式(t3)给出的结构的峰面积的比例的方法]
[0187]
在对调色剂颗粒中的thf不溶性物质进行
29
si
‑
nmr测量之后,通过将具有不同取代基和键合基团的多种硅烷组分进行曲线拟合,对调色剂颗粒进行向下述q1结构、q2结构、q3结构和q4结构的峰分离,并且由峰面积比来计算各组分的mol%。
[0188]
使用用于来自jeol ltd.的jnm
‑
ex400的excalibur for windows(注册商标)版本4.2(ex系列)软件来进行曲线拟合。从菜单图标点击“1d pro”并且加载测量数据。
[0189]
然后通过从菜单栏上的“命令(command)”中选择“曲线拟合功能”来进行曲线拟合。其实例在图3中给出。进行峰分割(peak resolution),从而使作为合成峰(b)与测量结果(d)之差的合成峰差(synthetic peak differential)(a)的峰最小。
[0190]
测定q1结构的面积、q2结构的面积、q3结构的面积和q4结构的面积,并且使用以下给出的公式来确定sq1、sq2、sq3和sq4。
[0191]
q1结构:(r1)(r2)(r3)sio
1/2
ꢀꢀꢀꢀꢀꢀꢀꢀ
式(2)
[0192]
q2结构:(r4)(r5)si(o
1/2
)2ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
式(3)
[0193]
q3结构:r6si(o
1/2
)3ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
式(4)
[0194]
q4结构:si(o
1/2
)4ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
式(5)
[0195][0196]
式(2)、(3)和(4)中的r1、r2、r3、r4、r5和r6表示与硅键合的有机基团、卤素原子、羟基或烷氧基。
[0197]
从化学位移值来鉴定硅烷单体,并且将有机硅聚合物的总峰面积取作来自对调色剂颗粒进行的
29
si
‑
nmr测量中的总峰面积中的q1结构的面积、q2结构的面积、q3结构的面积和q4结构的面积的总和。
[0198]
sq1 sq2 sq3 sq4=1.000
[0199]
sq1={q1结构的面积/(q1结构的面积 q2结构的面积 q3结构的面积 q4结构的面积)}
[0200]
sq2={q2结构的面积/(q1结构的面积 q2结构的面积 q3结构的面积 q4结构的面积)}
[0201]
sq3={q3结构的面积/(q1结构的面积 q2结构的面积 q3结构的面积 q4结构的面积)}
[0202]
sq4={q4结构的面积/(q1结构的面积 q2结构的面积 q3结构的面积 q4结构的面积)}
[0203]
由下式(t3)给出的结构的峰面积相对于有机硅聚合物的总峰面积为至少5.0%。即,在前面描述的测量方法中,表示由r
‑
sio
3/2
给出的结构的值为以上定义的sq3。其值为至少0.05。
[0204]
r
‑
si(o
1/2
)3ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(t3)
[0205]
以下给出q1结构、q2结构、q3结构和q4结构中的硅的化学位移值。
[0206]
q1结构的实例(r1=r2=
‑
oc2h5,r3=
‑
ch3):
‑
47ppm
[0207]
q2结构的实例(r4=
‑
oc2h5,r5=
‑
ch3):
‑
56ppm
[0208]
q3结构的实例(r6=
‑
ch3):
‑
65ppm
[0209]
在q4结构的情况下,硅的化学位移值如下。
[0210]
q4结构:
‑
108ppm
[0211]
<通过使用透射型电子显微镜(tem)观察调色剂颗粒截面来测量表层的平均厚度dav.和fran(表层的厚度)不超过5.0nm的arn线段的比例的方法>
[0212]
使用以下方法来进行调色剂颗粒截面的观察。
[0213]
在用于观察调色剂颗粒截面的具体方法中,将调色剂颗粒充分地分散在常温固化性环氧树脂中然后在40℃气氛中进行固化2天。使用装配有金刚石刀片的切片机从所得固化物切出薄片状样品。通过使用透射型电子显微镜(tem)(来自fei的tecnai tf20xt电子显微镜)以10,000x至100,000x的倍率放大该样品来观察调色剂颗粒截面。
[0214]
在本公开中,利用所使用的树脂和有机硅化合物中的原子的原子量之差,并且利用当原子量大时提供鲜明对比度的事实,来确认表层中有机硅聚合物的存在。使用四氧化钌的染色和使用四氧化锇的染色用于提供材料之间的对比度。
[0215]
dtem(由从tem显微照片获得的调色剂颗粒截面确定)在调色剂颗粒重均粒径(使用coulter counter通过以下给出的方法确定)的
±
10%的范围内的调色剂颗粒,是用于通过tem测量调色剂颗粒的表层的平均厚度dav.和测量fran(表层厚度)不超过5.0nm的arn线段的比例的目标调色剂颗粒。
[0216]
[由从tem显微照片获得的调色剂截面测量dtem的方法]
[0217]
使用长轴l(作为调色剂颗粒截面中的最大直径)与轴l90(穿过长轴l的中点并且与其垂直)之间的交点作为中心,将调色剂颗粒截面分割成32个均等的部分(参见图2)。即,
通过绘制穿过长轴l的中点并且在中点处以均等的交叉角(交叉角=11.25
°
)横穿截面的16条直线来形成从该中点至调色剂颗粒表面的32条线段。然后使用arn(n=1至32)作为从中心至调色剂颗粒表层的各线段(分割轴),使用ran作为各线段(分割轴)的长度,并且使用fran(n=1至32)作为线段arn上的表层的厚度。
[0218]
然后使用下式来确定由从tem显微照片获得的调色剂截面确定的dtem。
[0219]
dtem=(ra1 ra2 ra3 ra4 ra5 ra6 ra7 ra8 ra9 ra10 ra11 ra12 ra13 ra14 ra15 ra16 ra17 ra18 ra19 ra20 ra21 ra22 ra23 ra24 ra25 ra26 ra27 ra28 ra29 ra30 ra31 ra32)/16
[0220]
[调色剂颗粒的表层的平均厚度(dav.)的测量]
[0221]
使用以下方法来确定调色剂颗粒的表层的平均厚度(dav.)。首先,对于一个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒,使用以下方法来确定表层的平均厚度d(n)。
[0222]
d(n)=(分割轴上的表层的厚度在32个位置处的总和)/32=(fra1 fra2 fra3 fra4 fra5 fra6 fra7 fra8 fra9 fra10 fra11 fra12 fra13 fra14 fra15 fra16 fra17 fra18 fra19 fra20 fra21 fra22 fra23 fra24 fra25 fra26 fra27 fra28 fra29 fra30 fra31 fra32)/32
[0223]
为了求平均值,对于100个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒,确定调色剂颗粒表层的平均厚度d(n)(n=1至100),并且计算每1个调色剂颗粒的平均值并将其用作调色剂颗粒表层的平均厚度(dav.)。
[0224]
dav.={d(1) d(2) d(3) d(4) d(5)
·····
d(100)}/100
[0225]
[用于测量fran(表层厚度)不超过5.0nm的arn线段的比例的方法]
[0226]
首先,对一个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒进行操作,对于该调色剂颗粒,使用以下方法来确定fran(表层厚度)不超过5.0nm的arn线段的比例。
[0227]
[fran(表层厚度)不超过5.0nm的arn线段的比例]=({fran(表层厚度)不超过5.0nm的arn线段的数量}/32)
×
100
[0228]
对100个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒进行该计算,并且,对于这100个值,确定平均值并且将其用作调色剂颗粒的fran(表层厚度)不超过5.0nm的arn线段的比例。
[0229]
<用于测量表层中与核颗粒接触的无机细颗粒的数量的方法、和用于测量该数量为16个至30个的调色剂颗粒的比例的方法>
[0230]
在使用透射型电子显微镜(tem)观察调色剂颗粒的截面时,使用来自fei的tecnai tf20xt电子显微镜在200kv的加速电压下获取调色剂颗粒截面的明视场图像。然后,使用来自gatan,inc.的gif tridiem eels检测器,通过三窗口法(three window method)来获取si
‑
k端(99ev)的ef映射图像,并且鉴定各无机细颗粒的元素,并且对表层中与核颗粒接触的无机细颗粒的数量进行计数。对100个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒进行该测量,并且将其平均值用作在表层中接触核颗粒的无机细颗粒的数量。存在16个至30个在该表层中与核颗粒接触的无机细颗粒的调色剂颗粒的比例也类似地来确定。
[0231]
还使用该元素分析来确定无机细颗粒是否包含选自由钙元素和镁元素组成的组中的至少一者。
[0232]
<用于测量具有至少一个存在于核颗粒中并且不与表层接触的无机细颗粒的调色剂颗粒的比例的方法>
[0233]
存在于核颗粒中并且不与表层接触的无机细颗粒的观察可以按照上述用于测量表层中与核颗粒接触的无机细颗粒的数量的方法来进行。对100个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒进行该观察,以确定具有至少一个存在于核颗粒中并且不与表层接触的无机细颗粒的调色剂颗粒的比例。
[0234]
<无机细颗粒的一次粒径的数均粒径dm的测量>
[0235]
对于无机细颗粒的一次粒径的数均粒径dm,使用上述调色剂颗粒截面的观察中的放大图像来测量至少100个无机细颗粒的粒径,并且将其算术平均值确定为一次粒径的数均粒径dm。当形状为球状时,将其绝对最大长度记作粒径;当存在长径和短径时,将长径记作粒径。
[0236]
<在表层的八等分区域中的每一者中存在至少一个无机细颗粒的调色剂颗粒的比例>
[0237]
使用其中通过使用透射型电子显微镜(tem)观察调色剂颗粒截面来进行测量的、用于测量调色剂颗粒表层的平均厚度(dav.)和fran(表层厚度)不超过5.0nm的arn线段的比例的方法,将表层的八等分区域中的每一者中存在至少一个无机细颗粒的调色剂颗粒的比例确定为每100个dtem在调色剂颗粒重均粒径
±
10%的范围内的调色剂颗粒中在以下区域中的每一者中存在至少一个无机细颗粒的调色剂颗粒的比例:表层的夹在ar1和ar5之间的区域、表层的夹在ar5和ar9之间的区域、表层的夹在ar9和ar13之间的区域、表层的夹在ar13和ar17之间的区域、表层的夹在ar17和ar21之间的区域、表层的夹在ar21和ar25之间的区域、表层的夹在ar25和ar29之间的区域、和表层的夹在ar29和ar1之间的区域。
[0238]
<用于测量调色剂颗粒的重均粒径(d4)的方法>
[0239]
如下来确定调色剂颗粒的重均粒径(d4)。所使用的测量仪器为“coulter counter multisizer 3”(注册商标,beckman coulter,inc.),一种基于孔电阻法来操作并且装配有100μm口管的精密粒度分布测量仪器。使用所附专用软件,即,“beckman coulter multisizer 3版本3.51”(beckman coulter,inc.)来设定测量条件和分析测量数据。关于有效测量通道的数量,在25,000个通道中进行测量。
[0240]
通过将特级氯化钠溶解于去离子水中以提供1质量%的浓度来制备用于测量的电解质水溶液,并且,例如,可以使用“isoton ii”(beckman coulter,inc.)。
[0241]
在测量和分析之前,如下来设定专用软件。
[0242]
在专用软件的“改变标准操作方法(somme)”界面中,将控制模式中的总计数设定为50,000个颗粒;将测量次数设定为1次;并且将kd值设定为使用“标准颗粒10.0μm”(beckman coulter,inc.)获得的值。通过按下“阈值/噪声水平测量按钮”来自动地设定阈值和噪声水平。此外,将电流设定为1600μa;将增益设定为2;将电解质溶液设定为isoton ii;并且进行“测量后口管冲洗”的检查。
[0243]
在专用软件的“脉冲向粒径的转换设定”界面中,将元件间隔设定为对数粒径;将粒径元件设定为256个粒径元件;并且将粒径范围设定为2μm至60μm。
[0244]
具体测量过程如下。
[0245]
(1)将200ml上述电解质水溶液引入至multisizer 3专用的玻璃制250ml圆底烧杯中,将其置于样品架中并且以每秒24转进行使用搅拌棒的逆时针搅拌。通过专用软件的“口管冲洗”功能来预先除去口管内的污物和气泡。
[0246]
(2)将30ml电解质水溶液引入至玻璃制100ml平底烧杯中,并且向其中添加作为分散剂的、通过将“contaminon n”(包含非离子表面活性剂、阴离子表面活性剂和有机助洗剂的ph 7的精密测量仪器清洗用中性清洁剂的10质量%水溶液,来自wako pure chemical industries,ltd.)用去离子水三倍(质量)稀释来制备的0.3ml稀释液。
[0247]
(3)准备“超声分散系统tetora 150”(nikkaki bios co.,ltd.);这是一个电力输出为120w并且装配有配置为使相位偏移180
°
的两个振荡器(振荡频率=50khz)的超声分散器。将3.3l去离子水引入至超声分散器的水槽内并且向该水槽中添加2ml contaminon n。
[0248]
(4)将(2)中所述的烧杯设置在超声分散器上的烧杯固定孔中并且启动超声分散器。以使烧杯内的电解质水溶液的液面的共振状态最大化的方式来调整烧杯的高度位置。
[0249]
(5)在用超声波照射根据(4)设置的烧杯内的电解质水溶液的同时,将10mg调色剂颗粒以小等分量添加至电解质水溶液中并且进行分散。超声分散处理再持续60秒。在超声分散期间,将水槽中的水温适当地控制为10℃至40℃。
[0250]
(6)使用移液管,将在(5)中制备的含有分散的调色剂颗粒的电解质水溶液滴加至如(1)中所述的设置在样品架内的圆底烧杯中,调节以提供5%的测量浓度。然后,进行测量直至所测量的颗粒数量达到50,000个。
[0251]
(7)通过仪器所附的专用软件来分析测量数据并且计算重均粒径(d4)。当使用专用软件设定为图/体积%时,“分析/体积统计值(算术平均)”界面上的“平均直径”为重均粒径(d4)。
[0252]
实施例
[0253]
以下使用实施例和比较例来具体描述本公开,但是本公开不限于这些实施例和比较例或者受这些实施例和比较例限制。除非另有特别说明,否则实施例和比较例中所述的“份”在所有情况下均基于质量。
[0254]
<聚酯系树脂生产例>
[0255]
·
对苯二甲酸:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
11.1mol
[0256]
·
双酚a环氧丙烷2mol加合物:
ꢀꢀꢀꢀ
10.8mol
[0257]
将这些单体与酯化催化剂一起引入至高压釜中,并且高压釜安装有减压设备、水分离设备、氮气导入设备、温度测量设备和搅拌设备。在氮气气氛下降低压力的同时,通过常规方法在220℃下进行反应直至tg达到70℃以得到聚酯系树脂。重均分子量(mw)为8,200并且数均分子量(mn)为3,220。
[0258]
<调色剂1生产例>
[0259]
(用于补充添加的含有分散稳定剂的水系分散介质的制备)
[0260]
将以下材料引入至反应器中的350份去离子水中,并且,在用n2吹扫的同时,在60℃的温度下保温60分钟。
[0261]
磷酸钠
ꢀꢀꢀꢀ
14.0份
[0262]
10%盐酸
ꢀꢀꢀꢀ
7.0份
[0263]
在使用t.k.均质混合器(tokushu kika kogyo co.,ltd.)以12,000rpm搅拌的同时,将8.0份氯化钙溶解于20份去离子水中的氯化钙水溶液一次性全部引入,以制备含有磷酸钙的用于补充添加的水系分散介质。
[0264]
(造粒用水系分散介质的制备)
[0265]
将以下材料引入至反应器中的1,000份去离子水中,并且,在用n2吹扫的同时,在60℃的温度下保温60分钟。
[0266]
磷酸钠
ꢀꢀꢀꢀ
14.0份
[0267]
10%盐酸
ꢀꢀꢀꢀ
7.0份
[0268]
在使用t.k.均质混合器(tokushu kika kogyo co.,ltd.)以12,000rpm搅拌的同时,将8.0份氯化钙溶解于20份去离子水中的氯化钙水溶液一次性全部引入,以制备含有磷酸钙的造粒用水系分散介质。
[0269]
然后使用以下原料来制备聚合性单体组合物;将该步骤定义为溶解步骤。
[0270][0271]
通过使用磨碎机(nippon coke&engineering co.,ltd.)将这些原料分散3小时来制备聚合性单体组合物。然后将该聚合性单体组合物转移至另外的容器中并且在搅拌的同时在63℃下保持5分钟。然后添加20.0份聚合引发剂过氧化新戊酸叔丁酯(50%甲苯溶液)并且在搅拌的同时保持5分钟(溶解步骤)。
[0272]
将该聚合性单体组合物引入至造粒用水系分散介质中,并且在用高速搅拌器搅拌的同时造粒5分钟。然后添加30份用于补充添加的含有分散稳定剂的水系分散介质并且额外造粒15分钟(造粒步骤)。以12,000rpm进行造粒期间的搅拌,并且造粒温度为60℃。
[0273]
将高速搅拌器改变为螺旋桨式搅拌装置并且将内部温度升高至70℃。升高温度所需的时间为10分钟。在轻轻搅拌的同时进行反应5小时。ph为5.1。至此定义为反应1步骤。
[0274]
然后,通过添加1.0mol/l naoh水溶液在10分钟内将ph调节至8.0并且将容器内的温度升高至85℃。升高温度所需的时间为20分钟。然后使容器内保持在85℃下3.0小时。至此定义为反应2步骤。
[0275]
在完成反应2步骤之后,将回流冷凝管取下并且安装能够进行馏分回收的蒸馏设备。然后将容器内的温度升高至100℃。升高温度所需的时间为30分钟。然后将容器内的温度在100℃下保持5.0小时。该步骤完成时的ph为8.0。将从安装能够进行馏分回收的蒸馏设备之后至完成在100℃下5.0小时的保持时间定义为蒸馏步骤。将保持温度指定为蒸馏温
度,并且将保持时间指定为蒸馏时间。在该步骤期间除去残余单体和其它溶剂。
[0276]
在蒸馏步骤之后,进行冷却至30℃,将稀盐酸添加至容器内以使ph降低至1.5并且溶解分散稳定剂,并且进行过滤。在过滤后不取出所得滤饼,并且通过添加700份去离子水和再次过滤来进行洗涤。
[0277]
然后,在过滤后将滤饼取出并且在30℃下真空干燥1小时。
[0278]
使用风力分级机来切割粗粉和细粉。将由此获得的颗粒指定为调色剂颗粒1。将调色剂颗粒1自身指定为调色剂1。调色剂颗粒1的生产条件和配方在表1、2
‑
1和2
‑
2中给出,并且调色剂1的物性在表3中给出。
[0279]
<调色剂2至6和8至22生产例>
[0280]
除了使用表1、2
‑
1和2
‑
2中示出的生产条件和配方以外,根据调色剂1生产例来生产调色剂2至6和8至22。所得调色剂2至6和8至22的物性在表3中给出。
[0281]
<调色剂7生产例>
[0282]
(用于补充添加的含有分散稳定剂的水系分散介质的制备)
[0283]
将以下材料引入至反应器中的350份去离子水中,并且,在用n2吹扫的同时,在60℃的温度下保温60分钟。
[0284]
氢氧化钠
ꢀꢀꢀ
12.0份
[0285]
10%盐酸
ꢀꢀꢀ
5.0份
[0286]
在使用t.k.均质混合器(tokushu kika kogyo co.,ltd.)以12,000rpm搅拌的同时,将6.0份氯化镁溶解于20份去离子水中的氯化镁水溶液一次性全部引入,以制备含有氢氧化镁的用于补充添加的包含分散稳定无机细颗粒的水系介质。
[0287]
(造粒用水系分散介质的制备)
[0288]
将以下材料引入至反应器中的1,000份去离子水中,并且,在用n2吹扫的同时,在60℃的温度下保温60分钟。
[0289]
氢氧化钠
ꢀꢀꢀ
12.0份
[0290]
10%盐酸
ꢀꢀꢀ
5.0份
[0291]
在使用t.k.均质混合器(tokushu kika kogyo co.,ltd.)以12,000rpm搅拌的同时,将6.0份氯化镁溶解于20份去离子水中的氯化镁水溶液一次性全部引入,以制备含有氢氧化镁的造粒用水系介质。
[0292]
从造粒步骤之后,除了使用表1、2
‑
1和2
‑
2中示出的生产条件和配方以外,根据调色剂1生产例来生产调色剂7。所得调色剂7的物性在表3中给出。
[0293]
<调色剂23生产例>
[0294]
(粘结剂树脂细颗粒分散液1的制备)
[0295]
将80.0份苯乙烯、18.7份丙烯酸丁酯和1.3份作为用于提供羧基的单体的丙烯酸混合并且溶解。向该溶液中添加4.0份十二烷基苯磺酸钠混合在150份去离子水中的水溶液并且进行分散。在缓慢搅拌10分钟的同时,还添加0.3份过硫酸钾混合在10份去离子水中的水溶液。在用氮气置换之后,在70℃下进行乳液聚合6小时。在完成聚合之后,将反应液冷却至室温并且添加去离子水以获得固体浓度为20.0质量%并且基于体积的中值粒径为0.2μm的粘结剂树脂细颗粒分散液1。
[0296]
(聚酯树脂颗粒分散液的制备)
[0297]
将装配有冷凝器、温度计、水滴加装置和锚搅拌叶片的带夹套的3升反应槽(bj
‑
30n,tokyo rika kikai co.,ltd.)在水循环式恒温槽中保持在40℃。将160.0份乙酸乙酯和100.0份异丙醇的混合溶剂引入至该反应槽中;向其中引入300.0份无定形聚酯树脂(对苯二甲酸与环氧丙烷改性的(2mol加合物)双酚a的缩合物,mw=7,800,tg=70℃,酸值=8.0mg koh/g);并且通过使用三一电机(three
‑
one motor)以150rpm搅拌来溶解而获得油相。将14.0份10.0质量%氨水溶液经5分钟的滴加时间滴加至经搅拌的油相中,进行混合10分钟,然后,以每分钟7.0份的速度滴加900.0份去离子水以诱导相转变并且提供乳液。
[0298]
然后将800.0份获得的乳液和700.0份去离子水引入至2升回收瓶中,并且将其通过插入的捕集球安装在装有真空控制单元的蒸发器(tokyo rika kikai co.,ltd.)中。在使回收瓶旋转的同时,在60℃热水浴上进行加热并且将压力降低至7kpa以除去溶剂,同时注意避免暴沸。当溶剂回收量达到1,100.0份时,恢复至常压;将回收瓶进行水冷却以获得分散液。未从所得分散液检测到溶剂气味。该分散液中的聚酯树脂细颗粒的基于体积的中值粒径为130nm。添加去离子水以将固体浓度调整至20.0质量%,并且将其指定为聚酯树脂细颗粒分散液。
[0299]
(着色剂细颗粒分散液的制备)
[0300]
·
铜酞菁颜料(颜料蓝15:3):
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
100.0份
[0301]
·
阴离子表面活性剂、十二烷基苯磺酸钠:
ꢀꢀ
16.0份
[0302]
·
去离子水:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
384.0份
[0303]
将上述物质混合并且溶解,并且使用ultimizer高压冲击式分散机(hjp30006,sugino machine limited)对其进行分散60分钟,以生产其中分散有着色剂的着色剂细颗粒分散液。着色剂细颗粒分散液中的着色剂细颗粒的基于体积的中值粒径为130nm,并且着色剂细颗粒浓度为20.0质量%。
[0304]
(脱模剂细颗粒分散液的制备)
[0305]
·
费
‑
托蜡(熔点:78℃):
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
100.0份
[0306]
·
阴离子表面活性剂、十二烷基苯磺酸钠
ꢀꢀ
16.0份
[0307]
·
去离子水:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
384.0份
[0308]
将前述各组分混合,并且使用压力喷出型均化器(gaulin均化器,gaulin co.)使脱模剂在120℃的内部温度下溶解。然后在5mpa的分散压力下分散处理120分钟,然后在40mpa下分散处理360分钟,此后进行冷却以获得脱模剂细颗粒分散液。该脱模剂细颗粒分散液中的细颗粒的基于体积的中值粒径为225nm。随后,添加去离子水以将固体浓度调整至20.0质量%。
[0309]
(树脂颗粒1的制备)
[0310][0311]
将这些组分引入至安装有温度计、ph计和搅拌器的3升反应器中,并且在25℃的温度下通过添加0.3mol/l硝酸使ph达到3.0。然后,在使用均化器(ultra
‑
turrax t50,ika japan kk)以5,000rpm分散的同时,添加130.0份氯化铝水溶液(0.3质量%)并且进行分散6分钟。
[0312]
然后使反应容器安装有搅拌器和夹套式电阻加热器,并且调整搅拌器旋转速度从而充分地搅拌浆料。在继续搅拌的同时,将温度以0.2℃/分钟的升温速度升高至温度为40℃,然后,在超过40℃之后,以0.05℃/分钟的升温速度升高至90℃,并且在90℃下进行加热处理步骤180分钟。然后使用冷却水将容器冷却至20℃。
[0313]
冷却后,使浆料通过开口为15μm的尼龙筛网以除去粗粉末;将硝酸添加至已通过筛网的树脂颗粒分散液中以将ph调节至6.0;然后进行使用抽吸器的真空过滤。将残留在滤纸上的树脂颗粒用手尽可能细地粉碎,然后在30℃的温度下将其引入至相对于调色剂的量为10倍的去离子水中。在搅拌和混合30分钟之后,再次进行使用抽吸器的真空过滤并且测量滤液的电导率。通过重复该过程来洗涤树脂颗粒1直至滤液的电导率达到不超过5μs/cm。
[0314]
使用湿式/干式整粒机将经洗涤的树脂颗粒细粉碎,然后在35℃烘箱中真空干燥36小时以获得树脂颗粒。
[0315]
(树脂颗粒分散液)
[0316]
将400.0份去离子水引入至反应容器中。向其中添加表面活性剂(十二烷基苯磺酸钠)和金属盐(氯化铝六水合物),从而提供1.0
×
100质量%的表面活性剂浓度和40.0mmol/l的金属离子浓度。向其中添加100.0份树脂颗粒,并且在25℃的温度下使用均化器(ultra
‑
turrax t50,ika japan kk)以5,000rpm进行分散6分钟。然后使用1.0n氢氧化钠水溶液来将ph调整至9.0并且得到树脂颗粒分散液。
[0317]
然后,将100.0份树脂颗粒分散液量取至反应容器中并且在搅拌的同时将温度升高至70℃。向其中添加使用1mol/l naoh水溶液调整至ph 9.0的18.0份己基三乙氧基硅烷的水解溶液,并且进行搅拌240分钟以进行缩合步骤。
[0318]
然后使用kiriyama滤纸(no.5c,孔径=1μm)来过滤以从滤液中分离颗粒。将获得的颗粒用100份去离子水进一步洗涤并且在25℃下真空干燥24小时以获得调色剂颗粒23。调色剂颗粒23原样用作调色剂23。获得的调色剂23的物性在表3中给出。
[0319]
<调色剂24生产例>
[0320]
<粘结剂树脂颗粒分散液2的制备>
[0321]
将89.5份苯乙烯、9.2份丙烯酸丁酯、1.3份作为用于提供羧基的单体的丙烯酸和
3.2份正月桂基硫醇混合并且溶解。向该溶液中添加1.5份neogen rk(dai
‑
ichi kogyo seiyaku co.,ltd.)在150份去离子水中的水溶液并且进行分散。在缓慢搅拌10分钟的同时,还添加0.3份过硫酸钾在10份去离子水中的水溶液。在用氮气置换之后,在70℃下进行乳液聚合6小时。在完成聚合之后,将反应液冷却至室温并且添加去离子水以获得固体浓度为12.5质量%并且基于体积的中值粒径为0.2μm的粘结剂树脂颗粒分散液2。粘结剂树脂颗粒分散液2中包含的树脂包含源自丙烯酸的羧基。
[0322]
<脱模剂分散液的制备>
[0323]
将100份脱模剂(山嵛酸山嵛酯,熔点:72.1℃)和15份neogen rk混合在385份去离子水中,并且通过使用jn100湿式喷磨机(jokoh co.,ltd.)分散约1小时来获得脱模剂分散液。脱模剂分散液的浓度为20质量%。
[0324]
<着色剂分散液的制备>
[0325]
通过将100份作为着色剂的铜酞菁颜料(颜料蓝15:3)和15份neogen rk混合至885份去离子水中并且使用jn100湿式喷磨机分散约1小时来获得着色剂分散液。
[0326]
然后,使用均化器(ultra
‑
turrax t50,ika)来分散265份粘结剂树脂颗粒分散液2、10份脱模剂分散液和10份着色剂分散液。在搅拌的同时,将容器内的温度调整至30℃并且通过添加1mol/l氢氧化钠水溶液将ph调整至8.0(ph调整1)。在30℃下搅拌的同时,经10分钟添加0.3份氯化钙溶解于10份去离子水中的水溶液作为聚集剂。在静置3分钟之后开始加热,并且将温度升高至50℃以进行聚集颗粒的生成。在该状态下,使用“coulter counter multisizer 3”(注册商标,beckman coulter,inc.)来测量聚集颗粒的粒径。当重均粒径达到6.5μm时,添加0.9份氯化钠和5.0份neogen rk以使颗粒生长停止。
[0327]
向其中添加1.0份氯化钙作为补充添加的金属化合物,然后添加14.0份有机硅化合物己基三乙氧基硅烷,通过添加1mol/l氢氧化钠水溶液调整至ph=9.0(ph调整2),然后加热至95℃。在95℃下保持搅拌并且进行有机硅化合物的水解和缩合的同时,进行聚集颗粒的熔合和球形化。当平均圆形度达到0.980时开始冷却;在冷却至85℃之后,通过添加1mol/l氢氧化钠水溶液来进行调整至ph=9.5(ph调整3);进行搅拌180分钟以进一步促进缩合;并且随后进行冷却以获得调色剂颗粒分散液。
[0328]
通过如下来获得调色剂滤饼:将盐酸添加至获得的调色剂颗粒分散液中以将ph调整至不超过1.5,在搅拌的同时放置1小时,然后在加压过滤器上对调色剂颗粒分散液进行固液分离。将所述调色剂滤饼通过用去离子水再浆化再次制成分散液,然后进行在上述过滤器上的固液分离。重复再浆化和固液分离直至滤液的电导率达到不超过5.0μs/cm,此后进行最终固液分离以获得调色剂滤饼。将获得的调色剂滤饼在flash jet dryer气流干燥机(seishin enterprise co.,ltd.)中干燥。干燥条件为:吹入温度为90℃并且干燥机出口温度为40℃,并且与调色剂滤饼的水含量相对应地将调色剂滤饼供给速度调整为使出口温度不偏离40℃的速度。使用基于附壁效应的多级分级机来切割细颗粒和粗颗粒以得到调色剂颗粒24。调色剂颗粒24原样用作调色剂24。获得的调色剂24的物性在表3中给出。
[0329]
[表1]
[0330][0331]
[表2
‑
1]
[0332][0333]
[表2
‑
2]
[0334][0335]
[表3]
[0336][0337]
*1:归属于具有式(t3)的结构的峰面积相对于有机硅聚合物的总峰面积的比例
[0338]
*2:fran不超过5.0nm的arn线段的比例
[0339]
*3:具有至少一个与核颗粒接触并且不与表层接触的无机细颗粒的调色剂颗粒的比例
[0340]
*4:在表层的八等分区域中的每一者中存在至少一个无机细颗粒的调色剂颗粒的比例
[0341]
*5:表层中存在的与核颗粒接触的无机细颗粒的数量为16个至30个的调色剂颗粒的比例
[0342]
<实施例1至18和比较例1至6>
[0343]
对调色剂1至24进行以下评价。调色剂1至24的评价结果在表4中给出。
[0344]
<显影辊成膜的评价>
[0345]
以下具体地描述用于评价显影辊成膜的方法和评价标准。
[0346]
对于图像形成设备,使用具有如图1中的结构的串联激光束打印机hp color laser jet enterprise cp4525dn(hewlett
‑
packard)的改造机、和改造盒。
[0347]
通过改变内部传动装置以提供320mm/sec的处理速度来对该改造机进行改造。此外,将产品调色剂从盒内部取出,使用鼓风机进行清洁,并且填装250g调色剂。将该调色剂盒在评价实施环境中放置24小时,然后将其安装在打印机的青色站中;使用安装在其它站的虚拟盒(dummy cartridge)进行图像输出试验。
[0348]
通过重复每打印两张暂停一分钟的过程来输出50,000张调色剂承载水平为0.3mg/cm2的半色调图像,并且使用以下方法进行图像评价。
[0349]
(显影辊成膜的评价标准)
[0350]
通过显影辊表面的目视检查和通过图像的评价来评价显影辊成膜。
[0351]
在打印25,000张和50,000张之后,对于是否在打印的半色调图像的1%打印图像区域和非打印图像区域发生浓淡不均匀,进行目视评价。然后用空气吹扫显影辊的表面上的调色剂,并且观察显影辊的表面。
[0352]
a:在图像中未发生浓淡不均匀并且也不存在显影辊表面的成膜。
[0353]
b:在图像中未发生浓淡不均匀,但是在显影辊表面看到一些成膜。
[0354]
c:在显影辊表面看到成膜并且在图像中发生轻度的浓淡不均匀。
[0355]
d:在显影辊表面看到成膜并且在图像中发生显著的浓淡不均匀。
[0356]
<起雾的评价>
[0357]
为了评价起雾,在30.0℃/湿度80.0%rh环境中进行与显影辊成膜的评价中相同的图像输出试验,并且使用以下方法进行评价。
[0358]
(起雾的评价标准)
[0359]
在输出25,000张和50,000张之后,将改造机和改造盒在30.0℃/80.0%rh环境中放置三天。在放置期之后,输出具有白底区域的图像,并且通过使用如使用“reflectometer model tc
‑
6ds”(tokyo denshoku co.,ltd.)测量的评价纸的白度与输出图像的白底区域的白度之差计算起雾浓度(%)来评价图像起雾。通过将小数点后第二位四舍五入来确定起雾浓度。将绿光滤光器用作滤光器。
[0360]
a:起雾浓度不超过0.5%
[0361]
b:起雾浓度为至少0.6%且不超过1.5%
[0362]
c:起雾浓度为至少1.6%且不超过2.5%
[0363]
d:起雾浓度为至少2.6%
[0364]
<定影性能的评价>
[0365]
使用如上所述的改造机和改造盒在常温、常湿(25℃/50%rh)环境中操作,使用以5℃步长变化的定影温度来形成实心图像(调色剂承载水平:0.40mg/cm2)。使用普通纸(信纸尺寸xerox 4200纸,xerox corporation,75g/m2)作为转印材料。
[0366]
使用kimwipes(s
‑
200,crecia co.ltd.)将定影图像在75g/cm2的负荷下摩擦10次,并且基于摩擦前后的浓度下降率变得小于5%的温度和摩擦前后的浓度下降率变得小于10%的温度来评价低温定影性。使用反射浓度计(产品名:rd918,macbeth corporation)来测量图像浓度。
[0367]
(评价标准)
[0368]
a:不超过140℃
[0369]
b:145℃
[0370]
c:150℃
[0371]
d:至少155℃
[0372]
[表4]
[0373][0374]
虽然已经参考示例性实施方案描述了本发明,但是应当理解的是,本发明不限于公开的示例性实施方案。所附权利要求的范围要符合最宽泛的解释,从而涵盖所有此类修改以及等同的结构和功能。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。