1.本发明涉及药物检测技术领域,具体涉及一种佩兰及其制剂的质量控制方法、香豆素含量的测定方法及其应用。
背景技术:
2.佩兰为菊科植物佩兰eupatorium fortunei turcz.的干燥地上部分,具有芳香化湿,醒脾开胃,发表解暑之功效,用于湿浊中阻,脘痞呕恶、口中甜腻、口臭、多涎、暑湿表症,头胀胸闷,作为一种中医常用芳香化湿醒脾药,用药历史悠久。佩兰的化学成分包括挥发油、黄酮类、生物碱类、甾醇、脑苷脂类及其他成分,要确保佩兰的临床疗效,需要对其药效成分进行整体质量控制。
3.目前,《中国药典》2015版一部下佩兰项下尚未收载采用高效液相色谱法测定其成分含量或进行特征图谱控制的方法,并且现有技术中多是使用hplc对佩兰药材进行检测鉴别,而实际中,为了使用方便,往往将佩兰药材制成标准汤剂或配方颗粒进行使用,而标准汤剂或配方颗粒为佩兰药材水提后的有效成分,要想鉴别标准汤剂或配方颗粒的真伪或对其质量进行把控,也需要对其进行鉴别检测,但实际上,现有的适用于佩兰药材的hplc检测方法对于标准汤剂和配方颗粒的检测则不适用,而且,hplc液相方法还存在灵敏度低,耗时时间长,分离度低等问题。因此,亟需提供一种适用于佩兰标准汤剂和配方颗粒的、更为高效的检测方法。
技术实现要素:
4.因此,本发明要解决的技术问题在于克服现有技术中的检测方法不适用于佩兰标准汤剂和配方颗粒,耗时长、分离度低的缺陷,从而提供一种佩兰及其制剂的质量控制方法、香豆素含量的测定方法及其应用。
5.为此,本发明提供一种佩兰及其制剂的质量控制方法,包括获得佩兰及其制剂的特征图谱,所述特征图谱中包括1号峰、2号峰、3号峰和4号峰4个共有特征峰,3号峰为香豆素峰,以3号峰为参照物峰,其余峰的相对保留时间在规定值的
±
10%之内,规定值为:1号峰的相对保留时间为0.38,2号峰的相对保留时间为0.79,4号峰的相对保留时间为1.32。
6.进一步的,所述的佩兰及其制剂的质量控制方法,具体包括以下步骤:
7.制备供试品溶液:取佩兰样品制成供试品溶液;
8.具体可以为:取佩兰样品,加入水或有机溶剂进行提取,取溶液,即为供试品溶液;
9.制备对照药材参照物溶液:取佩兰对照药材制成对照药材参照物溶液;
10.具体可以为:取佩兰对照药材加水煎煮,然后取溶液,干燥后加入水或有机溶剂进行提取,取溶液,得到对照药材参照物溶液;
11.制备对照品参照物溶液:以香豆素为对照品制成对照品参照物溶液;
12.具体可以为:在香豆素中加入有机溶剂,得到对照品参照物溶液;
13.色谱条件:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以体积分数为0.08-0.12%的磷酸水溶液为流动相b,进行梯度洗脱,检测波长为274-278nm,柱温为33-37℃,流速为0.38-0.42ml/min;梯度洗脱程序具体为:0-10min,流动相a:流动相b的体积比为5%:95%
→
10%:90%;10-18min,流动相a:流动相b的体积比为10%:90%
→
22%:78%;18-23min,流动相a:流动相b的体积比为22%:78%
→
100%:0;32-40min,流动相a:流动相b的体积比为100%:0
→
5%:95%;
14.测定法:取对照药材参照物溶液、对照品参照物溶液和供试品溶液,分别注入超高效液相色谱仪,进行测定,得到对照特征图谱。
15.具体的,取对照药材参照物溶液、对照品参照物溶液和供试品溶液0.5μl-2.0μl,分别注入超高效液相色谱仪,进行测定,分别得到对照药材参照物溶液的液相色谱图、对照品参照物溶液的液相色谱图和供试品溶液的液相色谱图;
16.利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品参照物溶液以及对照药材参照物溶液的液相色谱图进行数据匹配,得到对照特征图谱。进一步地,所述佩兰样品为佩兰标准汤剂冻干粉、佩兰配方颗粒、佩兰饮片或佩兰药材。
17.进一步地,供试品溶液的具体制备过程为:精密称定佩兰样品0.1-1.0g,然后精密加入甲醇20ml,称定重量,超声或回流处理20-40min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液;
18.对照药材参照物溶液的具体制备过程为:精密称定佩兰对照药材0.1-1.0g,加水15-45ml,煎煮20-50min,离心,取上清液减压旋干,然后精密加入甲醇,称定重量,超声或回流处理20-40min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液;
19.对照品参照物溶液的具体制备过程为:精密称定香豆素对照品,加入甲醇,制成每1ml溶液中含10-70μg香豆素的对照品参照物溶液。
20.进一步地,供试品溶液的具体制备过程为:精密称定佩兰样品0.2g,然后精密加入甲醇20ml,称定重量,超声处理30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液;
21.对照药材参照物溶液的具体制备过程为:精密称定佩兰对照药材1.0g,加水30ml,煎煮35min,离心,取上清液减压旋干,然后精密加入甲醇20ml,称定重量,超声处理30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液;
22.对照品参照物溶液的具体制备过程为:精密称定香豆素对照品,加入甲醇,制成每1ml溶液中含50μg香豆素的对照品参照物溶液。
23.进一步地,磷酸水溶液的体积分数为0.1%,检测波长为276nm,柱温为35℃,流速为0.4ml/min。
24.本发明还提供一种佩兰及其制剂的香豆素含量的测定方法,包括如下步骤:
25.制备供试品溶液:取佩兰样品,加入水或有机溶剂进行提取,取溶液,即为供试品溶液;
26.制备对照品参照物溶液:在香豆素中加入有机溶剂,得到对照品参照物溶液;
27.色谱条件:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以体积分数为0.08-0.12%的磷酸水溶液为流动相b,进行梯度洗脱,检测波长为274-278nm,柱温为33-37℃,流速为0.38-0.42ml/min;梯度洗脱程序具体为:0-10min,流动
相a:流动相b的体积比为5%:95%
→
10%:90%;10-18min,流动相a:流动相b的体积比为10%:90%
→
22%:78%;18-23min,流动相a:流动相b的体积比为22%:78%
→
100%:0;32-40min,流动相a:流动相b的体积比为100%:0
→
5%:95%;
28.测定法:分别吸取对照品溶液和供试品溶液,注入超高效液相色谱仪,测定峰面积并计算香豆素含量。
29.进一步地,供试品溶液的具体制备过程为:精密称定佩兰样品0.2g,然后精密加入甲醇20ml,称定重量,超声处理30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液;所述佩兰样品为佩兰标准汤剂冻干粉、佩兰配方颗粒、佩兰饮片或佩兰药材;
30.对照品参照物溶液的具体制备过程为:精密称定香豆素对照品,加入甲醇,制成每1ml溶液中含50μg香豆素的对照品参照物溶液。
31.进一步地,磷酸水溶液的体积分数为0.1%,检测波长为276nm,柱温为35℃,流速为0.4ml/min。
32.本发明还提供一种上述的方法在佩兰及其制剂的质量检测及质量控制中的应用。
33.标准汤剂冻干粉:对标准汤剂进行减压浓缩后进行冷冻干燥所得到的冻干粉,其成分与标准汤剂保持一致。
34.本发明技术方案,具有如下优点:
35.1.本发明提供的佩兰及其制剂的质量控制方法,色谱条件适用于对经过水提、浓缩等工艺制备得到的佩兰药物制剂,如佩兰标准汤剂及佩兰配方颗粒,所用洗脱梯度更针对大极性成分,所得图谱峰形及分离度均较好,可以对佩兰标准汤剂、佩兰配方颗粒及佩兰饮片、药材进行多成分定性分析以及特定成分香豆素的定量分析,因此可以深入认识到佩兰的药效物质基础从药材到配方颗粒的工艺传递情况,保证物质基础的一致性,有利于整体评价从药材到配方颗粒的加工过程的合理性、科学性。
36.2.本发明提供的佩兰及其制剂的质量控制方法,供试品溶液、对照药材溶液的提取方法及对照品溶液的配制方法,能够同时满足定性和定量测定的需求,可以很好的控制佩兰药材、饮片、标准汤剂、配方颗粒的内在质量,保证产品从原料、中间体到成品的物质均一性和稳定性,确保佩兰临床疗效。
37.3.本发明提供的佩兰及其制剂超高效液相检测方法,可同时定性、定量检测,即可控制产品质量,又大大节省检测成本;方法操作简单,精密度高、稳定性好、重复性好、准确度高、分析速率快。
附图说明
38.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
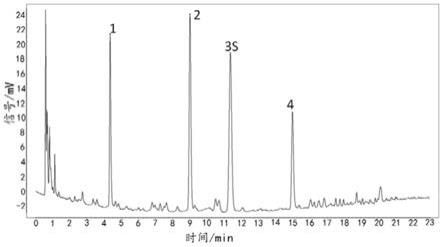
39.图1是本发明实施例1中供试品溶液超高效液相色谱图;
40.图2是本发明实施例1中得到对照特征图谱;
41.图3为本发明实施例2中供试品溶液的超高效液相色谱图;
42.图4为本发明实施例3中供试品溶液的超高效液相色谱图;
43.图5为本发明实施例4中供试品溶液的超高效液相色谱图;
44.图6为本发明实验例1中佩兰标准汤剂冻干粉延迟性谱图;
45.图7为本发明实验例2中香豆素含量测定检测波长的选择;
46.图8为本发明实验例2中香豆素峰纯度的检测;
47.图9为本发明实验例2中5.3专属性部分佩兰标准汤剂冻干粉供试品溶液的液相色谱图;
48.图10为本发明实验例2中5.3专属性部分香豆素对照品溶液的液相色谱图;
49.图11为本发明实验例2中5.3专属性部分甲醇阴性对照溶液的液相色谱图;
50.图12为本发明实验例2中5.3专属性部分辅料糊精阴性对照溶液的液相色谱图;
51.图13为本发明实验例2中5.4线性部分香豆素的标准曲线。
具体实施方式
52.提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
53.实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
54.以下实施例和实验例中用到的材料如下:
55.佩兰对照药材(批号:121113-201303,中国食品药品检定研究院)
56.香豆素对照品(批号:x-013-171216,纯度98.0%,成都瑞芬思生物科技有限公司);
57.18批佩兰药材或饮片供试品来源如表1所示:
58.表1
59.[0060][0061]
乙腈(merck)、磷酸(fisher scientific)为色谱纯;水为蒸馏水(屈臣氏);其他试剂均为分析纯。
[0062]
实施例1
[0063]
一种佩兰及其制剂特征图谱的建立方法
[0064]
标准汤剂冻干粉制备:分别取上述批号为190513-537000-01、190513-511600-02、190613-443600-03的佩兰药材,除去杂质,喷淋少量清水润湿药材(不能有存水浸泡药材),闷润30min,将闷软的佩兰药材放入剁刀式切药机内,饮片切成10-15mm的段,阴干,即得佩兰饮片;
[0065]
分别取18批次的佩兰饮片浸泡30min,煎煮二次,一煎加入饮片量12倍水,煎煮20min,二煎加入饮片量9倍水,煎煮10min,取佩兰标准汤剂滤液,使用真空减压浓缩设备,温度设置为50℃,浓缩至料液比约为1:1,得浓缩液,将浓缩液放置于托盘中,冷冻干燥,约30小时后,取出,研细,即得到18个批次的标准汤剂冻干粉。
[0066]
制备供试品溶液:
[0067]
精密称定佩兰标准汤剂冻干粉0.2g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到标准汤剂供试品溶液。
[0068]
制备对照药材参照物溶液:精密称定佩兰对照药材1.0g,加水30ml,煎煮35min,离心,取上清液减压旋干,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液。
[0069]
制备对照品参照物溶液:精密称定香豆素对照品,加入甲醇,制成每1ml溶液中含50μg香豆素的对照品溶液。
[0070]
色谱条件:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂(acquity beh shield rp 18,柱长为10cm,柱内径为2.1mm,粒径为1.7μm),以乙腈为流动相a,以体积分数为0.1%的磷酸水溶液为流动相b,进行梯度洗脱,检测波长为276nm,柱温为35℃,流速为0.4ml/min;梯度洗脱程序具体为:0-10min,流动相a:流动相b的体积比为5%:95%
→
10%:90%;10-18min,流动相a:流动相b的体积比为10%:90%
→
22%:78%;18-23min,流动相a:流动相b的体积比为22%:78%
→
100%:0;32-40min,流动相a:流动相b的体积比为100%:0
→
5%:95%;
[0071]
测定法:分别取对照药材参照物溶液、对照品参照物溶液和18批次的供试品溶液1.0μl,分别注入超高效液相色谱仪,进行测定,分别得到对照药材参照物溶液的液相色谱图、对照品参照物溶液的液相色谱图和供试品溶液的液相色谱图(图1);
[0072]
利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品参照物溶液以及对照药材参照物溶液的液相色谱图进行数据匹配,得到对照特征图谱(图
2)。具体的,使用《中药色谱指纹图谱相似度评价系统》(2012.1版本),以s1(即以批号为190513-537000-01的佩兰药材制备得到的标准汤剂冻干粉)液相图谱为参照图谱,按中位数计算,得出佩兰标准汤剂冻干粉对照特征图谱,并与对照药材参照物溶液和对照品参照物溶液液相色谱图进行共有峰的标识,共标识4个共有峰,其中峰3为香豆素峰。
[0073]
根据中药指纹图谱技术的要求对得到的18批佩兰标准汤剂冻干粉的特征图谱的相似度和相对保留时间进行分析。分析结果如下:
[0074]
1)相似度
[0075]
使用《中药色谱指纹图谱相似度评价系统》(2012.1版本),计算18批佩兰标准汤剂冻干粉的特征图谱与对照特征图谱的相似度,其结果见表2。
[0076]
表2相似度计算结果
[0077][0078]
小结:18批佩兰标准汤剂冻干粉特征图谱与对照特征图谱的相似度为0.946~1.000,均在0.90以上,表明多批次佩兰标煎汤剂冻干粉特征图谱满足相似度分析要求。
[0079]
2)相对保留时间
[0080]
对得到的18批佩兰标准汤剂特征图谱进行数据处理,得到相对保留时间数据,其结果如表3所示。
[0081]
表3 18批佩兰标准汤剂冻干粉相对保留时间表
[0082][0083][0084]
小结:由18批佩兰标准汤剂冻干粉特征图谱可知,各特征峰相对保留时间差异较小,均在
±
10%范围内,符合质量控制要求,选择相对保留时间的平均值作为测定值;规定值为:0.38(峰1)、0.79(峰2)、1.00(峰3s)、1.32(峰4),允许误差:
±
10%。
[0085]
实施例2
[0086]
取批号为190513-537000-01的佩兰药材,分别制备相应的饮片、标准汤剂冻干粉和配方颗粒,饮片及标准汤剂冻干粉的制备方法同实施例1;配方颗粒的制备方法:取佩兰饮片,煎煮二次,一煎加15倍水,沸腾(100℃)提取60min;二煎加12倍水,沸腾提取60min,取滤液,在温度为70℃以下、真空度为-0.09mpa的条件下减压浓缩,浓缩至相对密度1.03-1.10(60℃),然后喷雾干燥,加入辅料糊精(加入量为制成量的30%)干法制粒,得到配方颗粒。
[0087]
制备供试品溶液:
[0088]
精密称定佩兰标准汤剂冻干粉0.2g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到标准汤剂供试品溶液;
[0089]
精密称定佩兰配方颗粒0.2g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到配方颗粒供试品溶液;
[0090]
精密称定佩兰饮片0.2g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到饮片供试品溶液;
[0091]
精密称定佩兰药材0.2g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到药材供试品溶液;
[0092]
色谱条件:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂(acquity beh shield rp 18,柱长为10cm,柱内径为2.1mm,粒径为1.7μm),以乙腈为流动相a,以体积分数为0.1%的磷酸水溶液为流动相b,进行梯度洗脱,检测波长为276nm,柱温为35℃,流速为0.4ml/min;梯度洗脱程序具体为:0-10min,流动相a:流动相b的体积比为5%:95%
→
10%:90%;10-18min,流动相a:流动相b的体积比为10%:90%
→
22%:78%;18-23min,流动相a:流动相b的体积比为22%:78%
→
100%:0;32-40min,流动相a:流动相b的体积比为100%:0
→
5%:95%;
[0093]
测定法:分别不同的供试品溶液1.0μl,分别注入超高效液相色谱仪,进行测定,分别得到不同的供试品溶液的液相色谱图,如图3所示。
[0094]
实施例3
[0095]
取批号为190513-511600-02的佩兰药材,分别制备相应的饮片、标准汤剂冻干粉和配方颗粒,饮片及标准汤剂冻干粉的制备方法同实施例1,配方颗粒的制备方法同实施例4。
[0096]
精密称定佩兰标准汤剂冻干粉0.1g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)40min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到标准汤剂供试品溶液;
[0097]
精密称定佩兰配方颗粒0.1g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)40min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到配方颗粒供试品溶液;
[0098]
精密称定佩兰饮片0.1g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,
频率40khz)40min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到饮片供试品溶液;
[0099]
精密称定佩兰药材0.1g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)40min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到药材供试品溶液;
[0100]
色谱条件:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂(acquity beh shield rp 18,柱长为10cm,柱内径为2.1mm,粒径为1.7μm),以乙腈为流动相a,以体积分数为0.08%的磷酸水溶液为流动相b,进行梯度洗脱,检测波长为274nm,柱温为33℃,流速为0.38ml/min;梯度洗脱程序具体为:0-10min,流动相a:流动相b的体积比为5%:95%
→
10%:90%;10-18min,流动相a:流动相b的体积比为10%:90%
→
22%:78%;18-23min,流动相a:流动相b的体积比为22%:78%
→
100%:0;32-40min,流动相a:流动相b的体积比为100%:0
→
5%:95%;
[0101]
测定法:分别不同的供试品溶液0.5μl,分别注入超高效液相色谱仪,进行测定,分别得到不同的供试品溶液的液相色谱图,如图4所示。
[0102]
实施例4
[0103]
取批号为190613-443600-03的佩兰药材,分别制备相应的饮片、标准汤剂冻干粉和配方颗粒,饮片及标准汤剂冻干粉的制备方法同实施例1,配方颗粒的制备方法同实施例4。
[0104]
精密称定佩兰标准汤剂冻干粉1.0g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)20min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到标准汤剂供试品溶液;
[0105]
精密称定佩兰配方颗粒1.0g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)20min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到配方颗粒供试品溶液;
[0106]
精密称定佩兰饮片1.0g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)20min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到饮片供试品溶液;
[0107]
精密称定佩兰药材1.0g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)20min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到药材供试品溶液;
[0108]
色谱条件:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂(acquity beh shield rp 18,柱长为10cm,柱内径为2.1mm,粒径为1.7μm),以乙腈为流动相a,以体积分数为0.12%的磷酸水溶液为流动相b,进行梯度洗脱,检测波长为278nm,柱温为37℃,流速为0.42ml/min;梯度洗脱程序具体为:0-10min,流动相a:流动相b的体积比为5%:95%
→
10%:90%;10-18min,流动相a:流动相b的体积比为10%:90%
→
22%:78%;18-23min,流动相a:流动相b的体积比为22%:78%
→
100%:0;32-40min,流动相a:流动相b的体积比为100%:0
→
5%:95%;
[0109]
测定法:分别取不同的供试品溶液2.0μl,分别注入超高效液相色谱仪,进行测定,
分别得到不同的供试品溶液的液相色谱图,如图5所示。
[0110]
实验例1
[0111]
1.延迟性试验
[0112]
取佩兰标准汤剂冻干粉(批号:190513-537000-01)供试品溶液,按实施例1所述方法色谱条件进样,记录2倍流动相洗脱时间结果见图6。
[0113]
小结:图4结果表明,在23min之后没有明显的滞后峰出现,此色谱方法符合分析要求。
[0114]
2.系统适用性试验
[0115]
取佩兰标准汤剂冻干粉(批号:190513-537000-01)供试品溶液,按实施例1所述方法色谱条件进样,以香豆素峰为参照,进行系统适用性试验,其结果见表4。
[0116]
表4系统适用性试验结果
[0117][0118][0119]
小结:香豆素峰理论板数、拖尾因子及重复性均符合分析要求。
[0120]
3.方法学验证
[0121]
3.1精密度
[0122]
3.1.1重复性
[0123]
取按照实施例1所述方法得到的佩兰标准汤剂冻干粉(批号:190513-537000-01)6份,按实施例1所述方法测定,获得其特征图谱,以3号峰为参照峰,计算其相对峰面积和相对保留时间。并计算rsd。结果如表5、6所示。
[0124]
表5佩兰重复性考察保留时间及相对保留时间表
[0125]
[0126]
表6佩兰重复性考察峰面积及相对峰面积表
[0127][0128][0129]
小结:根据重复性考察结果,各特征峰的相对保留时间rsd在0.1%范围内,相对峰面积的rsd在1.6%~2.8%范围内,表明该特征图谱的重复性较好。
[0130]
3.1.2中间精密度
[0131]
采用waters uplc h-class,tuv检测器,取按照实施例1所述方法得到的佩兰标准汤剂冻干粉(批号:190513-537000-01)6份,按实施例1所述方法测定,获得其特征图谱,以3号峰为参照峰,计算其相对峰面积和相对保留时间。并计算rsd。结果如表7、表8所示。
[0132]
表7佩兰中间精密度考察保留时间及相对保留时间表
[0133][0134]
表8佩兰中间精密度考察峰面积及相对峰面积表
[0135][0136][0137]
小结:采用waters uplc h-class,tuv检测器获得的特征图谱中,各特征峰的中间精密度相对保留时间及相对峰面积均满足分析要求。
[0138]
3.2稳定性
[0139]
取按照实施例1所述方法得到的佩兰标准汤剂冻干粉(190513-537000-01),按实施例1所述方法制备供试品溶液,分别于0、2、4、6、8、10、12、24h时按实施例1方法进行测定,获得其特征图谱,以3号峰为参照峰,计算其相对峰面积和相对保留时间。并计算rsd。结果如表9、10所示。
[0140]
表9稳定性保留时间和相对保留时间表
[0141][0142]
表10稳定性相对峰面积表
[0143][0144][0145]
小结:稳定性实验结果可知,各特征峰的相对保留时间rsd在0.1%-0.2%范围内,相对峰面积的rsd在0.5%-1.1%范围内,表明样品溶液在24h内特征图谱中各成分较稳定。
[0146]
实施例5
[0147]
一种佩兰及其制剂的香豆素含量的测定方法
[0148]
佩兰饮片及标准汤剂冻干粉的制备方法同实施例1;
[0149]
制备佩兰配方颗粒的制备方法同实施例2。
[0150]
制备供试品溶液:
[0151]
精密称定佩兰标准汤剂冻干粉0.2g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到标准汤剂供试品溶液;
[0152]
精密称定佩兰配方颗粒0.2g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到配方颗粒供试品溶液;
[0153]
精密称定佩兰饮片0.2g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到饮片供试品溶液;
[0154]
精密称定佩兰药材0.2g,然后精密加入甲醇20ml,称定重量,超声处理(功率250w,频率40khz)30min,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过,取续滤液,得到药材供试品溶液;
[0155]
制备对照品溶液:精密称定香豆素对照品,加入甲醇,制成每1ml溶液中含50μg香豆素的对照品溶液。
[0156]
色谱条件:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂(acquity beh shield rp 18,柱长为10cm,柱内径为2.1mm,粒径为1.7μm),以乙腈为流动相a,以体积分数为0.1%的磷酸水溶液为流动相b,进行梯度洗脱,检测波长为276nm,柱温为35℃,流速为0.4ml/min;梯度洗脱程序具体为:0-10min,流动相a:流动相b的体积比为5%:95%
→
10%:90%;10-18min,流动相a:流动相b的体积比为10%:90%
→
22%:78%;18-23min,流动相a:流动相b的体积比为22%:78%
→
100%:0;32-40min,流动相a:流动相b的体积比为100%:0
→
5%:95%;
[0157]
测定法:分别取对照品溶液和供试品溶液1μl,分别注入超高效液相色谱仪,测定峰面积,按照外标一点法计算含量,计算公式如下:
[0158][0159]
其中,w
样
为供试品溶液的峰面积;
[0160]
c
对
为对照品参照物溶液的浓度(mg/ml);
[0161]
v
对
为对照品参照物溶液的进样体积(μl);
[0162]
w
对
为对照品参照物溶液的峰面积;
[0163]
c
样
为供试品溶液的浓度(g/ml);
[0164]
v
样
为供试品溶液的进样体积(μl);
[0165]
水分为供试品中的含水率。
[0166]
结果如表11所示:
[0167]
表11
[0168][0169]
实验例2
[0170]
1方法的原理
[0171]
本方法采用超高效液相色谱法对佩兰标准汤剂冻干粉中的香豆素进行测定,并利用外标一点法对香豆素进行定量。
[0172]
2实验条件的选择
[0173]
(1)波长的选择:按照实施例2所述的方法配制香豆素对照品溶液,利用二极管阵列检测器进行检测,确定香豆素的最大吸收波长276nm为检测波长,见图7。
[0174]
(2)确定的色谱条件:waters acquity beh shield rp18(2.1*100mm,1.7μm)色谱柱;以乙腈为流动相a,0.1%磷酸为流动相b,按表12进行梯度洗脱;检测波长为276nm,柱温35℃,流速0.4ml/min。
[0175]
表12梯度洗脱表
[0176][0177]
(3)峰纯度的检验:取佩兰标准汤剂冻干粉(批号:190513-537000-01),按照实施例2的方法制备供试品溶液,利用二极管阵列检测器对香豆素峰进行峰纯度检验,结果见表13、图8。
[0178]
表13佩兰标准汤剂冻干粉峰纯度表
[0179][0180]
结果显示,佩兰标准汤剂冻干粉的色谱图中,香豆素的峰纯角度小于纯度阈值2.781<39.743,表明该方法获得的香豆素峰纯度符合分析要求。
[0181]
3系统适用性
[0182]
按照实施例2的色谱条件,进行系统适用性试验,其结果见表14。
[0183]
表14系统适用性试验结果
[0184][0185]
结果显示,对照品香豆素理论板数为36277.2、拖尾因子为1.1、重复性rsd值为1.8,系统适用性符合分析要求。
[0186]
4供试品溶液的制备
[0187]
对提取溶剂的类型、溶液的浓度、提取方式和提取时间进行了考察。
[0188]
4.1提取溶剂的考察
[0189]
分别取佩兰标准汤剂冻干粉(批号:190513-537000-01)七份,每份0.2g,精密称定,置具塞锥形瓶中,分别精密加入适当溶剂(水、甲醇、乙醇、30%甲醇、70%甲醇、30%乙醇、70%乙醇)20ml,密塞,称定重量,超声处理(功率250w,频率40khz)30min,取出,放冷,再称定重量,分别用相应溶剂补足减失的重量,摇匀,滤过,精密吸取续滤液1.0μl,注入超高效液相色谱仪,按实施例2所述色谱条件测定,测定峰面积,计算香豆素的含量,结果见表15。
[0190]
表15不同提取溶剂的比较
[0191]
[0192]
小结:以上结果表明,甲醇和乙醇作为提取溶剂时香豆素含量较高,但乙醇为溶剂时的色谱图中各特征峰峰型较差,因此采用甲醇提取,其香豆素含量相对较高,峰型较好,故确定提取溶剂为甲醇。
[0193]
4.2提取浓度的考察
[0194]
分别取佩兰标准汤剂冻干粉(批号:190513-537000-01)0.1g、0.2g、0.5g、1.0g,精密称定,置具塞锥形瓶中,分别精密加入甲醇20ml,制成样品浓度是0.005g/ml、0.01g/ml、0.025g/ml、0.05g/ml的溶液,密塞,称定重量,超声处理(功率250w,频率40khz)30min,取出,放冷,称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。精密吸取续滤液1.0μl,注入超高效液相色谱仪,按正文色谱条件测定,测定峰面积,计算香豆素的含量。结果见表16所示。
[0195]
表16提取浓度的比较
[0196][0197]
小结:以上结果表明,采用0.005g/ml和0.01mg/ml的浓度提取均可以将佩兰标准汤剂冻干粉中的香豆素提取充分,考虑到液相信号响应程度,浓度大,信号相应值也越大,因此确定取样量为0.2g,提取溶剂量为20ml。
[0198]
4.3提取时间的考察
[0199]
分别称取佩兰标准汤剂冻干粉(批号:190513-537000-01)四份,每份0.2g,精密称定,置具塞锥形瓶中,分别精密加入甲醇20ml,密塞,称定重量,超声处理(功率250w,频率40khz)一定时间(20min、30min、40min),取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得,精密吸取续滤液1.0μl,注入超高效液相色谱仪,按正文色谱条件测定,测定峰面积,计算香豆素的含量,结果见表17。
[0200]
表17提取时间的比较
[0201][0202]
小结:以上结果表明,不同超声时间所获得的供试品溶液中香豆素含量差异不大,考虑到佩兰标准汤剂冻干粉的提取效率及提取含量差异,因此确定提取时间为30min。
[0203]
4.4提取方式的考察
[0204]
分别称取佩兰标准汤剂冻干粉(批号:190513-537000-01)两份,每份0.2g,精密称定,置具塞锥形瓶中,分别精密加入甲醇20ml,密塞,称定重量,采用不同的提取方式[超声处理(功率250w,频率40khz)、加热回流]提取30min,取出,放冷,称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得,精密吸取续滤液1.0μl,注入超高效液相色谱仪,按正文色谱条件测定,测定峰面积,计算香豆素的含量,结果见表18。
[0205]
表18不同提取方式的比较
[0206][0207]
小结:以上结果表明,不同提取方式所获得的供试品溶液中香豆素的含量差异不大,从实验操作的简单性考虑,确定采用超声处理作为供试品溶液制备的提取方式。
[0208]
5方法学验证
[0209]
5.1准确度
[0210]
取香豆素对照品适量,精密称定,加甲醇制成每1ml含14.881μg的溶液。取已知含量的佩兰标准汤剂冻干粉(190513-537000-01,香豆素含量为0.179%)平行三组共6份,每份约0.1g,精密称定,分别按1:0.5、1:1比例精密加入香豆素对照品溶液5ml(0.0744065mg香豆素)和甲醇15ml、香豆素对照品溶液10ml(0.148813mg香豆素)和甲醇10ml,余下操作同实施例2供试品溶液制备方法。
[0211]
取香豆素对照品适量,精密称定,加甲醇制成每1ml含17.826μg的溶液。取已知含量的佩兰标准汤剂冻干粉(190513-537000-01,香豆素含量为0.179%)平行三组共3份,每份约0.1g,精密称定,按照1:1.5比例精密加入香豆素对照品溶液15ml(0.267393mg香豆素)和甲醇5ml,余下操作同正文供试品溶液制备方法,按照下式计算回收率,结果见表19。
[0212][0213]
表19回收率试验结果表
[0214]
[0215][0216]
小结:回收率试验所测得的回收率范围为92.3%~108.0%,平均回收率为98.63%,rsd值为6.2,符合方法学验证回收率要求,表明利用该方法测得的结果准确。
[0217]
5.2精密度
[0218]
5.2.1重复性:取佩兰标准汤剂冻干粉(批号:190513-537000-01)6份,按实施例2方法测定含量,结果见表20。
[0219]
表20香豆素重复性试验
[0220][0221]
小结:重复性试验所测得佩兰标准汤剂冻干粉中香豆素平均含量为0.179%,其rsd为1.4%,符合方法学验证重复性要求。
[0222]
5.2.2中间精密度:不同分析人员在不同时间利用另一台waters uplch-class液相色谱仪(tuv检测器)进行中间精密度试验。取佩兰标准汤剂冻干粉(批号:190513-537000-01)6份,按实施例2方法进行测定含量,结果见表21。
[0223]
表21中间精密度试验结果表
[0224][0225]
小结:中间精密度试验所测得佩兰标准汤剂冻干粉中的香豆素平均含量为0.180%,rsd值为1.6%,与重复性试验的检测结果的rsd为0.3%,符合方法学验证精密度要求。
[0226]
5.3专属性
[0227]
取佩兰配方颗粒所使用的辅料(糊精)和甲醇适量,分别按照实施例2方法中供试品溶液的制备方法制备成阴性对照溶液;按实施例2所述方法制备佩兰标准汤剂冻干粉(批号:190513-537000-01)供试品溶液、香豆素对照品溶液,并按实施例2所述方法获得佩兰标准汤剂冻干粉供试品溶液、香豆素对照品溶液、阴性对照溶液的色谱图,其结果见图9-图12。
[0228]
5.4线性
[0229]
取香豆素对照品适量,加甲醇制成每1ml含0.108368mg的溶液,作为香豆素对照品
母液;分别精密吸取香豆素对照品母液1ml、3ml、5ml、7ml,置于10ml容量瓶中,加甲醇至刻度,摇匀,滤过,精密吸取续滤液1.0μl,注入超高效液相色谱仪,按实施例2所述方法测定香豆素色谱峰峰面积,以香豆素色谱峰的峰面积为纵坐标,香豆素的浓度为横坐标,观察是否呈线性,再用最小二乘法进行线性回归,求得回归方程为y=11,192,552.5928x-7,748.6885,r2=0.9998,其线性范围为0.010837-0.108368mg/ml。结果见表21、图13。
[0230]
表22香豆素标准曲线
[0231][0232]
5.5范围
[0233]
根据精密度、准确度和线性实验结果及多批次佩兰标准汤剂冻干粉中香豆素含量的结果,其范围为10.84mg/g~108.37mg/g。
[0234]
5.6耐用性
[0235]
5.6.1稳定性考察:取佩兰标准汤剂冻干粉(190513-537000-01),按实施例2方法制备供试品溶液,分别于0h、2h、4h、8h、10h、12h、24h时按实施例2方法进行测定,记录香豆素峰峰面积的变化情况,结果见表23。
[0236]
表23稳定性考察结果表
[0237][0238]
小结:由以上数据可知,24小时内香豆素峰的峰面积rsd值为0.6%,符合系统适用性试验要求。
[0239]
5.6.2不同柱温的考察:取佩兰标准汤剂冻干粉(批号:190513-537000-01),按实施例2所述的供试品溶液制备方法制备成供试品溶液,分别于不同柱温(33℃、35℃、37℃)下,按实施例2的方法进行测定,考察实验方法对于柱温的耐用性。结果如表24所示。
[0240]
表24不同柱温含量测定
[0241][0242]
小结:不同柱温下获得的色谱图,香豆峰的理论板数、拖尾因子、分离度均符合系统适用性要求;测得香豆素的含量rsd值为0.6%,基本符合系统适用性要求。表明该方法对柱温耐用性较好。
[0243]
5.6.3不同流速的考察:取佩兰标准汤剂冻干粉(批号:190513-537000-01),按实施例2供试品溶液的制备方法制备成供试品溶液,分别采用不同流速(0.38ml/min、0.40ml/min及0.42ml/min)按实施例2方法进行测定,考察实验方法对于流速的耐用性。结果如表25所示。
[0244]
表25不同流速含量测定结果表
[0245][0246]
小结:采用不同流速获得的色谱图,香豆素峰的理论板数、拖尾因子、分离度均符合系统适用性要求;测得香豆素的含量rsd值为0.9%,符合系统适用性要求。表明该方法对流速耐用性较好。
[0247]
5.6.4不同检测波长的考察:取佩兰标准汤剂冻干粉(批号:190513-537000-01),按实施例2供试品溶液的制备方法制备成供试品溶液,分别采用不同检测波长(274nm、276nm及278nm)按实施例2方法进行测定,考察实验方法对于检测波长的耐用性。结果如表26所示。
[0248]
表26不同检测波长含量测定结果表
[0249][0250]
小结:采用不同检测波长获得的色谱图,香豆素峰的理论板数、拖尾因子、分离度均符合系统适用性要求;测得香豆素的含量rsd值为0.9%,符合系统适用性要求。表明该方法对检测波长耐用性较好。
[0251]
5.6.5不同色谱柱的考察:取佩兰标准汤剂冻干粉(批号:190513-537000-01),按实施例2供试品溶液的制备方法制备成供试品溶液,分别采用不同色谱柱(beh c18、beh shield rp18及cortecs c18)按实施例2方法进行测定,考察实验方法对于不同色谱柱的耐用性。结果如表27所示。
[0252]
表27不同色谱柱含量测定结果表
[0253][0254]
小结:采用不同色谱柱获得的色谱图,香豆素峰的理论板数、拖尾因子、分离度均符合系统适用性要求;测得香豆素的含量rsd值为1.4%,符合系统适用性要求。表明该方法对色谱柱耐用性较好。
[0255]
5.6.6不同酸浓度的考察:取佩兰标准汤剂冻干粉(批号:190513-537000-01),按实施例2供试品溶液的制备方法制备成供试品溶液,流动相b分别采用不同酸浓度(0.08%、0.10%、0.12%磷酸溶液)按实施例2方法进行测定,考察实验方法对于不同酸浓度的耐用性。结果如表28所示。
[0256]
表28不同酸浓度含量测定结果表
[0257][0258]
小结:采用不同色谱柱获得的色谱图,香豆素峰的理论板数、拖尾因子、分离度均符合系统适用性要求;测得香豆素的含量rsd值为0.6%,符合系统适用性要求。表明该方法对酸浓度耐用性较好。
[0259]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。