1.本发明属于ndv的疫苗开发和以重组ndv为基础的其他生物制品制剂技术领域,具体涉及一种广谱型的新城疫病毒全基因组扩增引物和扩增方法。
背景技术:
2.新城疫(newcastle disease,nd),又称亚洲鸡瘟,是由新城疫病毒(newcastle disease virus,ndv)感染引起的禽的一种急性高度接触性传染病。新城疫病毒毒株众多,根据其毒力的不同,新城疫病毒可分为强毒株,中等毒力毒株和弱毒株。其中,强毒株ndv感染鸡只后,常常造成的毁灭性疾病,死亡率可高达90%以上,因此nd被世界动物卫生组织定为必须上报的疾病之一。
3.ndv属于副黏病毒科,禽副黏病毒属的成员。ndv虽然只有一个血清型,但基因型众多,并且随着病毒的不断进化,还有可能出现新的基因型。自从1999年第一株重组新城疫病毒被成功拯救出以来,目前全世界已有十余种不同毒株的新城疫病毒的反向遗传系统被建立。在建立新城疫反向遗传平台的过程中,完整无误的获得ndv的全基因组是公认的难点之一。其原因在于:一,基因组大。ndv是一种负链rna病毒,其基因组大小约15kb。负链rna病毒不能直接通过感染性rna来拯救,只能通过克隆全部的基因组序列构建质粒,并同时表达n蛋白,p蛋白,和l蛋白,形成rna聚合酶复合物来实现病毒的拯救。二,ndv病毒复制特征独特,ndv病毒复制基因组具有一个独特的规则,被称为“6原则”(rule of six),即新城疫病毒的基因组必须为6的倍数的时候,ndv才能被拯救成功,这一特征要求在构建ndv全长的时候,任何一个碱基的缺失或插入都可以导致病毒拯救的失败。
技术实现要素:
4.鉴于目前新城疫反向遗传学方法的缺点,本发明提供了一种广谱型的新城疫病毒全基因组扩增引物和扩增方法。
5.本发明采用如下技术方案:
6.一种广谱型的新城疫病毒全基因组扩增引物,包括以下引物:
7.part 1up:5
′‑
accaaacagagawtcbgtgag
‑3′
;
8.part 1down:5
′‑
aagaataccctccctgttgc
‑3′
;
9.part 2up:5
′‑
gcaacagggagrgtattcttytc
‑3′
;
10.part 2down:5
′‑
accaaacaragatttggtgaatg
‑3′
;
11.v1 up:5
′‑
agggagrgtattcttggccggcatggtcccagcctcc
‑3′
;
12.v1 down:5
′‑
gastctctgtttggtccctatagtgagtcgtattagcg
‑3′
;
13.v2 down:5
′‑
aaatctytgtttggtggccggcatggtcccagcctcc
‑3′
。
14.利用所述引物快速扩增新城疫病毒全基因组的方法,包括以下步骤:
15.s1、提取新城疫病毒的总rna,以所述总rna为模板,反转录合成cdna;
16.s2、以cdna为模板,在part 1up/part 1down和part 2up/part 2down的引导下用
rt
‑
pcr扩增得到part 1和part 2基因片段;
17.s3、将part 1和part 2基因片段连接、转化,挑取阳性克隆得到新城疫病毒全基因组序列。
18.进一步的,s2中,反应体系为:超纯水26μl,gxl polymerase 1μl,5
×
ps gxl buffer 10μl,2.5mm的dntp 10μl,part 1or 2up 1μl,part 1or 2down 1μl,模板3μl。
19.更进一步的,s2中,反应程序为:98℃,5min;98℃,10s,54℃,15s,68℃,8min,共30个循环;68℃,10min。
20.更进一步的,s3具体包括如下步骤:
21.(1)利用引物v1 up和v1 down对模板pbluescript sk 载体进行扩增,得到vector
‑
1载体;
22.(2)将part 1基因片段与vector
‑
1载体连接、转化,挑取阳性克隆得到vector1
‑
part1质粒;
23.(3)利用引物v1 up和v1 down对模板vector1
‑
part1质粒进行扩增,得到vector
‑
2载体;
24.(4)将part 1基因片段与vector
‑
2载体连接、转化,挑取阳性克隆并提取质粒,获得新城疫病毒全基因组序列。
25.更进一步的,步骤(1)和(3)中,反应体系均为:超纯水30μl,pfuμltraιι1μl,10
×
pfuμltraιιbuffer 5μl,2.5mm的dntp 10μl,v1 up 1μl,v1 down 1μl,100ng/μl的模板3μl。
26.更进一步的,步骤(1)和(3)中,反应程序均为:90℃,2min;92℃,20s,55℃,20s,68℃,3min,共35个循环;68℃,10min。
27.更进一步的,步骤(2)具体包括:将0.6μl part 1,7.4μl vector
‑
1和2μl 5
×
in
‑
fusion enzyme,混合均匀后,50℃,15min反应,得到重组真核表达载体i,将其转化入感受态细胞stble2中培养34
‑
36h,挑取单菌落,转接到含有羧苄西林的lb液体培养基中,30℃,180rmp的恒温摇床中培养12
‑
14h,然后提取质粒进行测序,获得连接正确的质粒命名为vector1
‑
part1。
28.更进一步的,步骤(4)具体包括:将1μl part 2,7μl vector
‑
2和2μl 5
×
in
‑
fusion enzyme,混合均匀后,50℃,15min反应得到重组真核表达载体ii,将其转化入感受态细胞stble2中培养34
‑
36h,挑取单菌落,转接到含有羧苄西林的lb液体培养基中,30℃,180rmp的恒温摇床中培养12
‑
14h,然后提取质粒进行测序,获得新城疫病毒全基因组序列。
29.与现有技术相比,本发明具有如下有益效果:
30.1、构建时间短。在以往的方案中,研究者通常将ndv的全基因组分为六个至十个基因片段进行扩增,然后再连接起来,实验步骤繁琐,实验周期长。而在本方法中,ndv的全基因组仅分为两段,只需要1轮到2轮的载体构建即可获得ndv的全基因组,大大缩短病毒全基因组构建所需时间。与以往ndv病毒拯救需要半年到一年,甚至两年的构建方案相比,该方法仅需要1个月的时间即可完成病毒全基因组的构建。
31.2、成功率高。众所周知,在载体构建过程中,出现基因缺失和插入的位点往往发生在目的基因和载体相连接的末端。该方案中,ndv的基因组基因和载体相连接的次数仅为一到两次,所以,该方法可以减少出现基因突变和缺失的概率,实现了高效率和高成功率的结
合。
32.3、适应于ndv不同的基因型。该方案在设计的时候,选取了众多ndv毒株的基因序列保守区域进行片段分割和引物设计,该方案适用于目前已知的大多数基因型的不同ndv毒株。虽然ndv只有一个血清型,但其基因型众多,并且不断的有新的毒株出现。本方法可以快速构建ndv新毒株的反向遗传平台,在ndv的疫苗开发和其他以ndv作为载体的生物制品的开发中具有重要的应用价值。
附图说明
33.图1为不同基因型的ndv毒株的保守区序列比较结果。
34.图2为本发明的ndv病毒全基因分析及分段构建ndv全基因组的流程示意图。
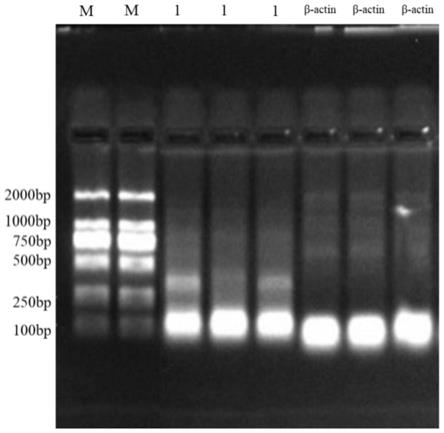
35.图3为本发明中ndv全基因组构建过程中各个基因片段扩增结果图,图中,m:1.5kb dna marker;f1:线性化的pbluescript sk( );f2:ndv part 1扩增结果;f3:ndv part 2扩增结果;f4:亚克隆vector1
‑
part1扩增结果;f5:全基因组质粒vector2
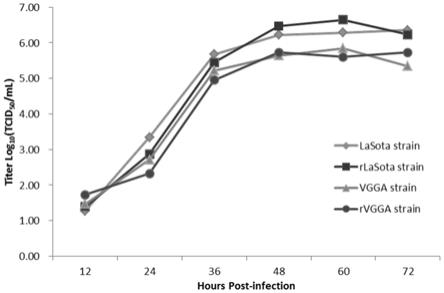
‑
part2扩增结果。
36.图4为本发明中所制备的重组病毒的免疫荧光检测结果图。
37.图5为本发明中重组病毒的一步生长曲线图。
具体实施方式
38.下面结合附图和具体实施例对本发明进行详细说明,但不应理解为本发明的限制。如未特殊说明,下述实施例中所用的技术手段为本领域技术人员所熟知的常规手段,下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
39.以下实施例及试验例中使用到的ndv由本实验室纯化保存提供。为了验证本研究所建立的快速构建重组ndv的方法的有效性,本研究选取了ndv的vgga avine和lasota株作为研究对象,分别构建了两者的反向遗传系统,拯救了两者的重组病毒。其中,经全基因测序,本研究的ndv lasota疫苗株的基因序列与genbank中新城疫lasota株(genbank accession no.jf950510.1)的序列一致,vgga avine株的序列与vgga avine株(genbank accession no.jf950510.1)的序列一致。f48e9株序列见(genbank accession no.mg456905)。辅助质粒pci
‑
n、pci
‑
p和pci
‑
l质粒通过将表达新城疫病毒lasota株的n、p和l基因(基因序列见genbank accession no.jf950510.1)连接入promega公司pci
‑
neo质粒构建完成。感受态细胞stble
‑
2购自上海唯地生物技术有限公司。gxl高保真polymerase购自宝生物(大连)有限公司。pfuultra high
‑
fidelity dna polymerase ad购自安捷伦有限公司。in
‑
fusion试剂盒购自clontech公司。病毒核酸提取试剂盒购自takara公司,微柱浓缩dna凝胶回收试剂盒和高纯度质粒小提中量提取试剂盒购自北京庄盟国际生物基因科技有限公司。其他常规试剂均为国产分析纯。
40.实施例1:新城疫病毒全基因组扩增策略及相应引物设计
41.本实施例中通过比对不同基因型ndv毒株的全基因组序列信息,分析ndv基因组中的保守区域,将ndv的基因组分两段扩增,设计出快速扩增和构建ndv全基因组cdna质粒的方案,具体包括如下步骤:
42.1、ndv病毒的基因组特征分析:
43.在ndv的反向遗传操作系统构建的过程中,ndv的全基因组构建入质粒是公认的难
点和重点,在ndv病毒是否能够被成功拯救中具有决定性的作用。为此,在以往的方案中,研究者通常将ndv的全基因组分为六个至十个基因片段进行扩增,然后再依次连接构建入质粒载体中,该方法实验步骤繁琐,实验周期长。为了解决这一难题,本研究通过比不同基因型的ndv毒株的全基因序列信息,以期建立一种可以应用于多种ndv基因型的ndv全基因组构建方法。为此,特选以下具有代表性的毒株进行分析:v4(genbank accession no.af217084);i
‑
2progenitor(genbank accession no.ay935500);b1
44.(genbank accession no.af309418);vgga(genbank accession no.eu289028);lasota(genbank accession no.ef440343);go/ch/lhlj/2/06(genbank accession no.kj607167);mukteswar(genbank accession no.jf950509);sh15(genbank accession no.ky247177);r2b(genbank accession no.jx316216);js/9/05/go(genbank accession no.fj430160);
45.herts/33(genbank accession no.ay741404);italien(genbank accession no.eu293914);lahore/25a/2015(genbank accession no.kx236101);dove/italy/2736/00(genbank accession no.gq429293);sweden_95
46.(genbank accession no.hq839733);fontana(genbank accession no.ay562988);na
‑
1(genbank accession no.dq659677);sweden/97(genbank accession no.gu585905);f48e9(genbank accession no.mg456905);hbnu/lsrc/f3(genbank accession no.kc246549);gd09
‑
2(genbank accession no.hq317394)等。
47.分析结果如图1所示,其中,ndv基因组的3
′
端前21个核苷酸比较保守,多为“accaaacagagaatccgtgag”,基因1型到3型的序列完全一致,4型之后的基因型序列中在13位由a突变为t和16位核苷酸由c突变为t。而ndv的基因组5
′
端的25个碱基全部为“accaaacagagaatccgtgag”,说明ndv的基因组的两段均比较保守。与此同时,通过序列的比对我们发现,ndv的基因组中7055位到7084位核苷酸序列相当保守,多为“gcaacagggagggtattctt”,仅有少数毒株存在单个碱基的突变,且多发生在该序列的中部,综合分析这三部分的序列特征。
48.2、ndv全基因组快速构建方案制定及相关引物设计
49.在明确ndv基因组的保守区域的基础上,本研究通过保守区的基因构成,长度大小,以及现有的高保真dna聚合酶的扩增能力等多重因素的基因上,将ndv基因组分为两个片段,大小分别为7084bp(或7090bp)和8102bp。具体方案详见图2。
50.本研究通过采用in
‑
fusion技术进行ndv全基因组载体构建,其引物设计需要目的基因和载体片段之间具有15bp的基因重复序列,具体的引物详见表1。
51.表1 pcr扩增引物序列
[0052][0053]
3、ndv病毒的全基因组的构建
[0054]
为了验证本研究所建立的快速ndv全基因组构建方法,本研究以实验室保存的ndv毒株,基因1型的ndv疫苗株vgga
‑
avine和基因2型疫苗株lasota及强度株f48e9为例,进行ndv的全基因组构建和反向遗传学方法建立,具体如下:
[0055]
(1)ndv part 1和part 2基因的rt
‑
pcr扩增
[0056]
取经过三次有限系稀释法纯化的ndv vgga
‑
avine、lasota及f48e9病毒原液为对象,以takara公司的病毒核酸提取试剂盒提取ndv的病毒rna。之后,以ndv病毒rna为模板,通过rt
‑
pcr法扩增目的序列part 1和part 2。其中,rt反应阶段,序列part 1的cdna扩增引物为part 1up;part 2的cdna扩增的特异性引物为part 2up;其它试剂和反应条件严格按照takara公司的primescript
tm ii high fidelity rt
‑
pcr kit进行cdna的合成。
[0057]
之后以part 1的cdna和part 2的cdna为模板,分别通过pcr反应扩增part 1和part 2基因,其中,扩增vgga
‑
avine和lasota采用的引物均为a型引物,因f48e9的3
′
的基因序列与前两者不同,在扩增f48e9的时候采用b型引物,具体反应体系和反应条件见表2和表3。
[0058]
表2 ndv part 1,2的pcr反应体系(50μl)
[0059][0060]
表3 ndv part 1,2的pcr扩增反应条件
[0061][0062][0063]
反应结束后,进行琼脂糖凝胶电泳并回收pcr扩增产物ndv part 1和part2(参照微柱浓缩dna凝胶回收试剂盒说明书),回收后送检测序,并放于
‑
20℃备用。
[0064]
本发明成功扩增到了ndv vgga株和lasota株的part 1和part 2基因,经基因测序表明part 1和part 2的基因大小分别为7084bp和8102bp。基因序列分别与genbank中的序列一致,没有碱基的缺失、突变和插入。
[0065]
(2)质粒vector 1和vector 2的扩增及载体构建
[0066]
a、首先,以pbluescript sk( )载体为模板,通过50μl的pcr反应体系进行vector 1载体扩增,所用引物为v1 up和v1 down,反应体系与反应条件表4和表5。
[0067]
表4 vector 1载体的pcr反应体系(50μl)
[0068][0069]
表5 vector 1载体片段扩增的反应条件
[0070][0071][0072]
b、in
‑
fusion方法构建重组真核表达载体vector1
‑
part1
[0073]
将已经测量过浓度的0.6μl ndv part 1,7.4μl vector
‑
1和2μl 5
×
in
‑
fusion enzyme,混合均匀后,50℃,15min反应后,得到重组真核表达载体i,置于4℃保存。随后将上述in
‑
fusion方法所连接的重组真核表达载体i转化入感受态细胞stble2中培养34
‑
36h。挑取单菌落,分别转接到含有羧苄西林的lb液体培养基中,30℃,180rmp的恒温摇床中培养12
‑
14h。然后提取质粒进行测序,获得连接正确的质粒命名为vector1
‑
part1。
[0074]
c、在获得vector1
‑
part1的基础上,以vector1
‑
part1质粒为模板,通过50μl的pcr反应体系进行vector 2载体扩增,所用引物为v1 up和v2 down,反应体系与反应条件表6和表7。
[0075]
表6 vector 2载体的pcr反应体系(50μl)
[0076][0077]
表7 vector2载体片段扩增的反应条件
[0078][0079][0080]
d、in
‑
fusion方法构建重组真核表达载体vector2
‑
part2
[0081]
将已经测量过浓度的1μl ndv part 2,7μl vector
‑
2和2μl 5x in
‑
fusion enzyme,混合均匀后,50℃,15min反应后,得到重组真核表达载体ii,置于4℃保存。随后将上述in
‑
fusion方法所连接的重组真核表达载体ii转化入感受态细胞stble2中培养34
‑
36h。挑取单菌落,分别转接到含有羧苄西林的lb液体培养基中,30℃,180rmp的恒温摇床中培养12
‑
14h。然后提取质粒进行测序,获得连接正确的质粒命名为vector2
‑
part2。其中vector2
‑
part2质粒为本发明中构建得到的含有ndv全基因组的质粒载体。
[0082]
pcr扩增及载体构建的结果见图3。
[0083]
实施例2:ndv病毒的拯救
[0084]
1、含有ndv全基因组的质粒vector2
‑
part2的细胞转染
[0085]
实验前将bhk
‑
21细胞传代至六孔板,待细胞状态良好时,弃去6孔板内培养基,向每个孔加入1.5ml opti
‑
mem无血清培养基,每孔加入表达t7 rna酶的痘病毒(mva
‑
t7病毒),静置于37℃培养箱备用。dna和脂质体预混合准备:离心管a:加入750μl opti
‑
mem无血清培养基和45μl lipofectamine 3000,涡旋振荡2~3s混合均匀,平均分装6管;离心管b:加入750μl opti
‑
mem无血清培养基和30μl lipofectamine 3000,平均分装6管,每管加入1μg的vector2
‑
part2,同时加入辅助质粒1μg pci
‑
n、0.5μg pci
‑
p和0.1μg pci
‑
l。将离心管
b中的混合试剂加入到离心管a中,室温静置15min。将复合物滴加到对应六孔板中,轻微晃动六孔板使复合物混合均匀。置于37℃、5%co2培养箱静置培养36
‑
48h。
[0086]
2、转染产物的鸡胚尿囊腔接种
[0087]
转染48小时后,用封口膜密封将6孔细胞板,转移到
‑
80℃低温冰箱中,反复冻融3次,液体移至离心管中,12000r离心2min,吸出上清,收获拯救的重组病毒。在鸡胚尿囊腔处寻找血管较少处进行标记,在超净工作台中用打孔器在标记处打一小孔,用注射器从打孔处进入向鸡胚注射0.1ml病毒,用石蜡严密封口,在37℃保温箱中培养。每天观察鸡胚状态并在24h内弃去死胚。培养96h后,将所有鸡胚置于4℃过夜,致死鸡胚,然后收集尿囊液。首先使用小镊子将壳膜撕开,露出气室,用移液管吸收透明的尿囊液。置于4℃保存备用。
[0088]
实施例3:重组ndv的生物学特性检测
[0089]
1、拯救病毒rlasota和rvgga的鉴定
[0090]
(1)重组病毒rlasota和rvgga的传代培养
[0091]
稀释病毒后,接种9至11日龄的spf鸡胚。使用照蛋器标记出鸡胚尿囊腔的位置,转入超净工作台中,使用打孔器在标记上打孔,将0.1~0.2ml的病毒注入鸡胚。用石蜡严密封口,转入37℃培养箱中培养。于48~96小时左右收获病毒,收获的重组病毒分别命名为rlasota和rvgga,第一代重组病毒经0.22μm滤膜过滤两次后,按同样方法再次进行鸡胚注射,进行三次传代。随后对第三代病毒进行ha测定。
[0092]
(2)重组病毒rlasota和rvgga的ha鉴定
[0093]
吸取50μl pbs缓冲液加入96孔血凝板中。在第一纵排孔中加入50μl尿囊液,吹打15至8次充分混合后,吸取50μl注入第二纵排孔。再次混合6至8次,随后将50μl液体添加到第三纵排孔中。依此类推,直到添加到第11纵排孔中,吹打混合,弃去50μl。第12纵排孔加入50μl pbs液做为阴性对照。每孔中加50μl1%鸡红细胞悬液,混匀,置于25℃培养箱中20~30分钟,观察结果。
[0094]
观察结果:鸡红细胞出现明显的凝集现象,说明本发明成功拯救出了重组新城疫病毒rlasota和rvgga。
[0095]
(3)rlasota和rvgga的间接免疫荧光法鉴定
[0096]
试验前一天,将df
‑
1细胞接种于12孔细胞培养板内,2ml/孔,用含有10%胎牛血清、1%抗生素的dmem培养基培养,于37℃培养箱过夜培养。待细胞状态良好时接种病毒rlasota和rvgga。37℃培养24h,细胞病变后,弃去培养液,用温热的pbs洗涤两次。然后用10%福尔马林溶液500μl室温固定15min,弃液,用pbs洗涤两次。加入100%无水乙醇500μl,置于
‑
20℃,5min以穿透细胞,用pbs洗涤三次。加入5%羊血清封闭,加入以稀释度为1:100的鼠抗ndv hn单抗(购自圣克鲁斯生物技术公司,货号sc
‑
53562)为一抗,37℃放置30min。以pbs洗涤三次,每次静置5min。再加入稀释度为1:200的alexa fluor标记的羊抗鼠血清(北京博奥森生物技术有限公司,货号bs
‑
0296g
‑
af555)为二抗,37℃放置30min,以pbs洗涤三次,每次静置5min。加入fluoromount
‑
g荧光保护剂,然后在倒置荧光显微镜下观察。
[0097]
结果如图4所示,将rlasota和rvgga感染df
‑
1细胞后,24h后通过免疫荧光的方法可以看出,df
‑
1细胞和ndv的抗体出现明显的结合反应而被染上红色的荧光,说明ndv病毒在df
‑
1细胞中成功表达。
[0098]
2、rlasota和rvgga的生物学特性检测
[0099]
(1)eid
50
的测定
[0100]
用pbs缓冲液连续稀释病毒,取稀释度为23、24、25、26、27的病毒分别接种于9~11日龄spf鸡胚各5枚,每枚鸡胚尿囊接种量为0.1ml,置于37℃恒温培养箱中培养,每天照蛋,弃掉24h内死亡的鸡胚,至第五天,将所有鸡胚置于4℃过夜后,收获每枚鸡胚的尿囊液,进行血凝实验,ha实验呈阳性时判断为感染,计算eid
50
结果。
[0101]
(2)tcid
50
的测定
[0102]
在离心管中将病毒液作连续10倍的稀释,取稀释度为101、102、103、104、105、106的病毒接种在96孔培养板中,每一稀释度接种一纵排,共8孔,每孔100μl,并向每孔加入100μl细胞悬液,将最后两个纵行设置为对照,向每个孔中加入100μl培养基和100μl细胞悬浮液,并且每天观察细胞病变状况并记录结果。计算tcid
50
的值。
[0103]
(3)mdt的测定
[0104]
将三代病毒用pbs缓冲液作连续倍比稀释,取稀释度为105、106、107、108、109的病毒分别接种9~11日龄spf鸡胚5只,每枚鸡胚接种量为0.1ml,置于37℃恒温培养箱中培养,每天早晚观察2次,连续7天,记录鸡胚死亡的时间,计算mdt的结果。
[0105]
表8重组病毒生物学特性鉴定
[0106][0107][0108]
结果如表8所示,经过eid
50
、tcid
50
和ha的检测结果可以看出rlasota和rvgga的病毒滴度和亲本毒株基本一致。其中rvgga的tcid50、eid50和ha测定结果表明rvgga比亲本毒株vgga株的增殖能力略微降低,但病毒滴度仍旧满足作为疫苗研发母本的要求。而同时mdt的检测结果可以看,rlasota和rvgga保持了亲代病毒的弱毒特征,甚至毒力稍弱于亲代毒株。
[0109]
3、重组病毒的生长曲线的检测
[0110]
病毒生长生长曲线的检测在6孔细胞培养板中进行,首先将df
‑
1细胞接种至6孔细胞培养板,每孔含有2
×
106个细胞。将moi值为0.01的重组病毒用无血清培养液稀释后,接种于df
‑
1细胞后,37℃孵育1小时。去除培养物后,细胞维持在含有1%的尿囊液的无血清培养基中培养96h。每24h收集上清液,96h后采用tcid
50
在df
‑
1细胞检测各个时间点的病毒滴度,绘制病毒生长曲线。
[0111]
结果如图5所示,表明本研究获得的重组病毒rlasota和rvgga的生长曲线与原病毒基本一致。结合病毒的其他生物学特征分析表明本研究构建的重组病毒保持了原有病毒的毒力和增殖特征,为开发以这些病毒为载体的基因工程疫苗或者其他生物制品奠定了基础。
[0112]
因f48e9为强毒株ndv病毒,尽管本研究成功的构建了其全长的基因组cdna,因实验条件所限,本研究未进行病毒的拯救工作。但是鉴于测序分析其基因组完全正确,说明其
病毒拯救是完全可行的。
[0113]
尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
[0114]
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。