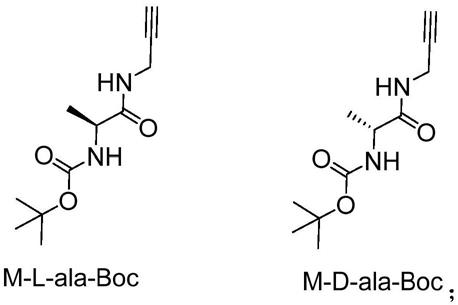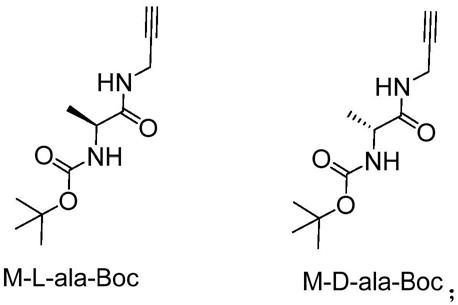
1.本发明涉及光学活性的高分子材料领域,具体涉及一种负载金属离子的单手性螺旋共聚取代聚炔及其制备方法和应用。
背景技术:
2.光学活性聚合物是科学家们一直关注的研究热点,通过不同的手段可以让聚合物的光学活性纯度提高,获得更加稳定的螺旋结构。除了在自然环境中,天然高分子及其所组成的动植物体都呈现出稳定的螺旋状结构,人工合成的螺旋聚合物一般来说都会采取调整聚合物主链或者是侧链构型使聚合物拥有手性构型。而聚炔是一种典型的线性共轭高分子,具有适当取代基的聚炔能够形成螺旋结构,左、右手螺旋取代聚炔呈镜像异构关系(chem.rev.2009,109,6102
–
6211)。与当前研究和应用较多的烯类聚合物相似,取代聚炔具有优异的侧基结构可设计性,易于功能化,能被用于构筑功能丰富的手性材料。其构象能在溶剂、添加剂等外部刺激下发生改变。
3.光学活性氨基酸是一种典型的旋光分子,广泛的存在于天然蛋白质中,也是最为廉价的一类手性分子,因此是仿生启迪人工螺旋聚合物合成的首选原料。在聚合物的侧基上引入氨基酸基团合成较强光学活性的单手性螺旋聚合物已经是高分子学家研究的重点之一,专利cn111410812a公开了一种氨基酸基螺旋聚硅烷红外吸收材料的制备方法,氨基酸提供的手性基团可以诱导高分子形成特定的螺旋结构,赋予了聚合物较强的光学活性。2003年masuda等人将氨基酸侧基引入聚炔的侧基上,成功合成了光学活性优异的油溶性氨基酸基取代聚炔(macromolecules 2003,36,3938
‑
3943),将手性氨基酸一对对映体共聚,呈现出的“大多数规则”类型的手性放大效应丰富了单手性螺旋聚合物种类。为单手性螺旋聚合物的制备提供了新的简单可实现的技术方案。
技术实现要素:
4.本发明的目的在于提供一种具有强光学活性的氨基酸基螺旋聚合物,丰富光学活性聚合物的种类。
5.为实现上述目的,本发明采用的技术方案是:
6.一种负载金属离子的单手性螺旋共聚取代聚炔,所述单手性螺旋共聚取代聚炔由如下结构式的聚合物与m形成配位结构:
[0007][0008]
其中,m包括cu
2
、ag
、au
3
、zn
2
或ni
2
中任一种;n表示聚炔的聚合度。
[0009]
本发明中通过单手性螺旋共聚取代聚炔负载重金属离子,在共聚取代聚炔的基础上再次将手性放大,具有较强的光学活性,比旋光度在25℃下绝对值高达1100
°
。
[0010]
本发明还提供所述的负载金属离子的单手性螺旋共聚取代聚炔的制备方法,其特征在于,包括如下步骤:
[0011]
步骤1,n
‑
叔丁氧羰基
‑
l
‑
丙氨酸或n
‑
叔丁氧羰基
‑
d
‑
丙氨酸与氯甲酸异丁酯、炔丙胺进行酰胺化反应制备得到取代炔单体m
‑
l
‑
ala
‑
boc或m
‑
d
‑
ala
‑
boc,其结构式如下:
[0012][0013]
步骤2,将m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc在催化剂条件下共聚合得到油溶性共聚物p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc,其结构式如下:
[0014][0015]
其中r表示m
‑
l
‑
ala
‑
boc的投料占比,n表示聚炔的聚合度;
[0016]
该步骤的反应式如下:
[0017][0018]
步骤3,将所述油溶性共聚物p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc溶于良溶剂中,与三氟乙酸反应后,再加入路易斯碱,得到水溶性共聚物p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2,其结构式如下:
[0019][0020]
该步骤的反应式如下:
[0021][0022]
步骤4,将所述水溶性共聚物p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2与含有金属离子的水溶液混合,得到所述负载金属离子的单手性螺旋共聚取代聚炔。
[0023]
本发明在含有手性氨基酸基团的共聚取代聚炔侧基中负载金属离子,形成单手性氨基酸基螺旋聚合物金属离子配合物,金属离子与氨基酸具有较强的配位能力,在金属离子的刺激作用下,螺旋聚炔的二级结构发生构象转变。在共聚取代聚炔“大多数规则”的基础上再次进行手性放大作用,得到强光学活性的氨基酸基螺旋聚合物,为单手性螺旋聚合物的制备提供了新的方法,同时丰富了光学活性聚合物的种类。
[0024]
优选地,当m为cu
2
时,所述单手性螺旋共聚取代聚炔则具有如下结构式:
[0025]
该步骤的反应式如下:
[0026][0027]
步骤2中,m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc的摩尔比为1.2
‑
9:1或1:1.2
‑
9。
[0028]
两种单体的摩尔量不能相等,否则会发生自消旋从而共聚物失去手性,但任一单体的比例含量也不宜过高,因此优选地,m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc的摩尔比为1.5
‑
9:1或1:9
‑
1.5。例如两者摩尔比为:1:9、1.5:8.5、2:8、2.5:7.5、3:7、3.5:6.5、4:6、、6.5:3.5、8:2、9:1等等。通过改变m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc的投料比,可使聚合物拥有相反的单手螺旋结构。
[0029]
步骤2中,共聚温度为10
‑
40℃,共聚时间为30min
‑
24h。
[0030]
步骤2中,催化剂物质的量是m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc的总摩尔量的0.1%
‑
10%。催化剂用量过低则所得共聚物的产率大幅下降;用量过高,聚合成本大幅提高,聚合
物中会残留未除去的催化剂,影响下一步反应。
[0031]
优选地,所述催化剂为有机铑配合物催化剂,进一步优选地,催化剂选自(nbd)rh
b
‑
(c6h5)4、rh[p(och3)3]2[b(c6h5)4]或rh[p(c6h5)3]2[b(c6h5)4]中任一种;在一些实施例中,催化剂为(nbd)rh
b
‑
(c6h5)4。
[0032]
步骤3中,所述良溶剂选自二氯甲烷、三氯甲烷、四氯甲烷、甲苯、二甲苯、四氢呋喃、n,n
‑
二甲基甲酰胺或n,n
‑
二甲基乙酰胺中的至少一种;所述路易斯碱选自碳酸钾、碳酸钠、醋酸钾、醋酸钠、碳酸氢钾、碳酸氢钠、磷酸钠或磷酸钾中至少一种。路易斯碱目的在于去除多余的三氟乙酸,其添加量为三氟乙酸摩尔量的1.5倍以上,确保三氟乙酸充分去除即可。
[0033]
步骤3中,所述三氟乙酸与油溶性共聚物p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc的摩尔比不低于1.5:1。三氟乙酸目的在于脱除油溶性共聚物中的叔丁氧羰基保护基团,如添加量不足,会导致保护基团脱除率有所降低。
[0034]
步骤4中,金属离子与所述水溶性共聚物p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的摩尔比为0.1
‑
2:1;这是由于若比例小于0.1,金属离子的含量不足以诱导整个聚合物主链形成螺旋结构,比例大于2,会造成多余的金属离子与水形成氢键,影响聚合物和金属络合物的溶解性,同时造成金属盐的浪费。
[0035]
由于金属离子更容易发生配位反应,因此本发明步骤3中所述金属离子包括铜离子、银离子、金离子、锌离子或镍离子中任一种。不同的金属离子种类将导致共聚物的螺旋类型和旋向发生改变。
[0036]
步骤4中,反应温度为10℃
‑
40℃,反应时间为3h
‑
24h。
[0037]
本发明还提供所述的负载金属离子的单手性螺旋共聚取代聚炔在滤光片中的应用。
[0038]
与现有技术相比,本发明具有以下有益效果:
[0039]
(1)本发明中的单手性螺旋共聚取代聚炔由于侧基手性基团和金属离子的加入,使不同比例的共聚物呈现出单手螺旋构型。通过改变m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc的投料比,可使共聚物拥有不同光学活性强度的单手螺旋结构;通过改变金属离子的种类,从而改变共聚物的螺旋类型和旋向。由于金属离子的存在,共聚取代聚炔呈现出更加显著的手性放大效应,提高了该共聚物的比旋光度。
[0040]
(2)本发明中采用的原料成本较低,且采用两步法得到水溶性共聚物,避免使用昂贵的水溶性铑催化剂,降低成本,利于工艺推广应用。
附图说明
[0041]
图1为实施例1中p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc的核磁共振氢谱图。
[0042]
图2为实施例1中p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc的cd和uv
‑
vis光谱图。
[0043]
图3为实施例1中p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc的比旋光度曲线图。
[0044]
图4为实施例1中p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc的ft
‑
ir光谱图。
[0045]
图5为实施例1中p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的cd和uv
‑
vis光谱图。
[0046]
图6为实施例1中p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的比旋光度曲线图。
[0047]
图7为实施例1中p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的ft
‑
ir光谱图。
[0048]
图8为实施例1中cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的cd和uv
‑
vis光谱图。
[0049]
图9为实施例1中cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的比旋光度曲线图。
[0050]
图10为实施例1中cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的的ft
‑
ir光谱图。
[0051]
图11为实施例12中ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的cd和uv
‑
vis光谱图。
[0052]
图12为实施例12中ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的比旋光度曲线图。
具体实施方式
[0053]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。本领域技术人员在理解本发明的技术方案基础上进行修改或等同替换,而未脱离本发明技术方案的精神和范围,均应涵盖在本发明的保护范围内。
[0054]
以下具体实施方式中所采用的原料、仪器均为市场所购,未经处理直接使用。
[0055]
实施例1
[0056]
第一步制备油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc,具体如下:将n
‑
叔丁氧羰基
‑
l
‑
丙氨酸或n
‑
叔丁氧羰基
‑
d
‑
丙氨酸与氯甲酸异丁酯和炔丙胺进行酰胺化反应,得到取代炔单体m
‑
l(ord)
‑
ala
‑
boc;m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc以投料比为6:4在总单体摩尔量1%的(nbd)rh
b
‑
(c6h5)4的作用下30℃进行溶液聚合24h,再经过后处理得到油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc;
[0057]
第二步制备水溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体如下:将p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc溶于二氯甲烷中,加入油溶性共聚物摩尔量1.5倍的三氟乙酸脱除boc保护基,在30℃反应24h。再加入碳酸钾除去三氟乙酸制得水溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2。
[0058]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2:将p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2溶解于去离子水中,再加入与共聚物单体单元比为1:1的铜离子水溶液,30℃下反应24h后,获得负载铜离子的单手性螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2。
[0059]
对实施例中得到的中间体和产物进行检测,检测结果如图1
‑
图10所示。
[0060]
图1为p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc的核磁共振氢谱图。从图中看出,1h nmr谱图中无杂质峰,各化学位移对应于单体结构上的每种氢,且峰面积与氢个数成正比,说明成功制得油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc。其中r=0.6时,ee%=r
‑
(1
‑
r)=0.2=20%。单体聚合的转化率为98.8%,几乎接近100%,说明共聚物组成与单体进料比几乎相同,可以认为发生了无规共聚。
[0061]
图2为p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc的cd和uv
‑
vis光谱图。均聚物的cd光谱在400nm处表现出镜像cd信号,这表明p
‑
l
‑
ala
‑
boc和p
‑
d
‑
ala
‑
boc呈相反方向的螺旋结构。均聚取代聚炔和ee%=80%的共聚取代聚炔的uv
‑
vis光谱在395nm处显示最大吸收,cd信号几乎没有变化。共聚物的cd信号与单体单元比呈非线性的关系,说明其中存在“大多数规则”(手性放大效应中的一种),而在ee%=20%的共聚取代聚炔,主吸收不是在395nm,而是在312nm,这表明20%的共聚取代聚炔比均聚取代聚炔的共轭程度低,因此,与单手过量螺旋构象相比,这些聚合物主链的π共轭片段减少,呈无规构象。说明ee%=20%的共聚取代聚炔是油溶性
取代聚炔中“大多数规则”服从程度最低。
[0062]
图3为p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc的比旋光度曲线图。在室温下,p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc的比旋光度在
‑
1100
°
至 1106
°
范围内。比旋光度与共聚物中单体单元的对映体过量百分比呈非线性关系。掺入10%的旋光异构体(ee%=80%)几乎不影响比旋光度,而掺入25%的异构体(ee%=50%)导致比旋光度变化大。在ee%=20%和ee%=0%之间的比旋光度差异相对较小。比旋光度的结果表明,共聚物的螺旋度与单体单元比呈非线性的关系。说明其中存在“大多数规则”,且ee%=20%的共聚取代聚炔是油溶性取代聚炔中“大多数规则”服从程度最低的。与图2结论一致。
[0063]
图4为p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc的ft
‑
ir光谱图。从红外光谱可知,不同投料比两种单体合成的p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
boc的红外谱图几乎一致,说明不同螺旋主链几何结构几乎没有变化。说明共聚只影响主链构象的变化,而不影响主链的构型。
[0064]
图5为p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的cd和uv
‑
vis光谱图。从图中可以看出,uv
‑
vis中300nm波长位置不随着掺入的旋光异构体量的变化而变化。不同投料比的p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的cd信号几乎为0,说明不同投料比p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的构象均为无规线团。
[0065]
图6为p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的比旋光度曲线图。通过旋光和cd,uv
‑
vis测试表征共聚物的构象。在室温下,p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的比旋度在 1
°
至 14
°
范围内。比旋光度最大的值不超过14,说明几乎没有旋光性,即无光学活性。
[0066]
图7为p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的ft
‑
ir光谱图。从红外光谱可知,水溶性取代聚炔p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2成功合成。同时,不同投料比的p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的螺旋构象几乎一致,且主链几何结构也几乎一致。说明不同投料比的共聚取代聚炔p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的二级结构均为无规线团结构。
[0067]
图8为cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的cd和uv
‑
vis光谱图。不同投料比的cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的cd光谱在275nm和335nm处对应地表现出镜像的cd信号。不同投料比cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的紫外吸收位置和cd出峰位置几乎没有变化,只是cd信号强度在随着ee%值的增大而减小。ee%=80%的cu
2
‑
p
‑
l
0.9
‑
co
‑
d
0.1
‑
ala
‑
nh2表现出与cu
2
‑
p
‑
l
‑
co
‑
ala
‑
nh2几乎相同的cd信号强度。而掺入60%的异构体(ee%=20%)才导致cd信号强度有显著的变化。与比旋光度的结果一致,再次说明存在“大多数规则”。同时,从紫外光谱可知,除了cu
2
‑
p
‑
l
0.5
‑
co
‑
d
0.5
‑
ala
‑
nh2构象为消旋螺旋,其他不同投料的cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2均为单手过量螺旋结构。将油溶性取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc进行脱保护处理、负载铜离子之后,得到的水溶性取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2的“大多数规则”服从程度明显提高,且表现出优异的光学活性,二级结构为单手螺旋结构。
[0068]
图9为cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的比旋光度曲线图。在室温下,cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的比旋度在
‑
1174
°
至 1134
°
范围内。比旋光度与共聚物中单体单元的对映体过量百分比呈非线性关系。掺入10%的旋光异构体(ee%=80%)几乎不影响比旋光度,而掺入60%的异构体(ee%=20%)导致比旋光度变化大。说明共聚物和cu
2
络合物服从“大多数规则”的服从程度高。比旋光度的结果表明,共聚物的螺旋度与共聚物中单体单元的对映体过量百分比呈非线性的关系。与图8结论一致。
[0069]
图10为cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的的ft
‑
ir光谱图。从红外光谱可知,不同投料比的cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2成功合成,同时cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的螺旋主链几何
结构几乎没有变化。1392cm
‑1处的聚炔衍生物主链上的c
‑
h的面内弯曲振动峰也几乎没有变化,也是侧基甲基上的c
‑
h的面内弯曲振动峰。在1392cm
‑1处的ir峰主要是由p
‑
l
‑
ala
‑
nh2主链上顺式
‑
顺式结构的形成引起的,可能部分是由顺式c=c含量的增加引起的。这意味着在螺旋主链中c=c的顺式含量不变,说明不同共聚比例不会影响主链的实际构型。聚合物和铜离子的相互作用力也几乎没有变化。
[0070]
本发明对实施例1中的每步产物都进行了表征,用ft
‑
ir定性地表征每步产物的结构,用cd和uv
‑
vis光谱表征共聚取代聚炔的二级结构和金属离子负载在共聚取代炔上的构象变化。
[0071]
对比例1
[0072]
第一步制备油溶性共聚取代聚炔p
‑
l
0.5
‑
co
‑
d
0.5
‑
ala
‑
boc,具体如下:将n
‑
叔丁氧羰基
‑
l
‑
丙氨酸或n
‑
叔丁氧羰基
‑
d
‑
丙氨酸与氯甲酸异丁酯和炔丙胺进行酰胺化反应,得到取代炔单体m
‑
l(or d)
‑
ala
‑
boc;m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc以投料比为5:5在总单体摩尔量1%的有机铑配合物催化剂的作用下30℃进行溶液聚合24h,再经过后处理得到油溶性共聚取代聚炔p
‑
l
0.5
‑
co
‑
d
0.5
‑
ala
‑
boc;
[0073]
第二步制备水溶性共聚取代聚炔p
‑
l
0.5
‑
co
‑
d
0.5
‑
ala
‑
nh2,具体如下:将p
‑
l
0.5
‑
co
‑
d
0.5
‑
ala
‑
nh2溶于二氯甲烷中,加入油溶性共聚物摩尔量1.5倍的三氟乙酸脱除boc保护基,在30℃反应24h。再加入碳酸钾除去三氟乙酸制得p
‑
l
0.5
‑
co
‑
d
0.5
‑
ala
‑
nh2。
[0074]
第三步制备cu
2
‑
p
‑
l
0.5
‑
co
‑
d
0.5
‑
ala
‑
nh2,将p
‑
l
0.5
‑
co
‑
d
0.5
‑
ala
‑
nh2溶解于去离子水中,再加入与共聚物单体单元比为1:1的铜离子水溶液,30℃下反应24h后,获得负载铜离子的单手性螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.5
‑
co
‑
d
0.5
‑
ala
‑
nh2。
[0075]
对最终得到的取代聚炔进行光学活性测试,结果由于m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc投料比为1:1,所以螺旋聚合物主链形成内消旋。故无光学活性,不能形成单手性螺旋聚合物。
[0076]
实施例2
[0077]
第一步制备油溶性共聚取代聚炔p
‑
l
0.9
‑
co
‑
d
0.1
‑
ala
‑
boc,具体如下:将n
‑
叔丁氧羰基
‑
l
‑
丙氨酸或n
‑
叔丁氧羰基
‑
d
‑
丙氨酸与氯甲酸异丁酯和炔丙胺进行酰胺化反应,得到取代炔单体m
‑
l(or d)
‑
ala
‑
boc;m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc以投料比为9:1在总单体摩尔量1%的(nbd)rh
b
‑
(c6h5)4催化剂的作用下30℃进行溶液聚合24h,再经过后处理得到油溶性共聚取代聚炔p
‑
l
0.9
‑
co
‑
d
0.1
‑
ala
‑
boc;
[0078]
第二步制备水溶性共聚取代聚炔p
‑
l
0.9
‑
co
‑
d
0.1
‑
ala
‑
nh2,具体如下:将p
‑
l
0.9
‑
co
‑
d
0.1
‑
ala
‑
nh2溶于二氯甲烷中,加入油溶性共聚物摩尔量1.5倍的三氟乙酸脱除boc保护基,在30℃反应24h。再加入碳酸钾除去三氟乙酸制得p
‑
l
0.9
‑
co
‑
d
0.1
‑
ala
‑
nh2。
[0079]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.9
‑
co
‑
d
0.1
‑
ala
‑
nh2,将p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2溶解于去离子水中,再加入与共聚物单体单元比为1:1的铜离子水溶液,30℃下反应24h后,获得负载铜离子的单手性螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.9
‑
co
‑
d
0.1
‑
ala
‑
nh2。
[0080]
各项检测结果实施例2与实施例1结果相似,只是实施例2中需要对映体纯度较高的单体进行聚合,需要的成本更高。
[0081]
实施例3
[0082]
第一步制备油溶性共聚取代聚炔p
‑
l
0.8
‑
co
‑
d
0.2
‑
ala
‑
boc,具体如下:将n
‑
叔丁氧羰基
‑
l
‑
丙氨酸或n
‑
叔丁氧羰基
‑
d
‑
丙氨酸与氯甲酸异丁酯和炔丙胺进行酰胺化反应,得到下式所示取代炔单体m
‑
l(or d)
‑
ala
‑
boc;m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc以投料比为8:2在总单体摩尔量1%的(nbd)rh
b
‑
(c6h5)4的作用下30℃进行溶液聚合24h,再经过后处理得到油溶性共聚取代聚炔p
‑
l
0.8
‑
co
‑
d
0.2
‑
ala
‑
boc;
[0083]
第二步制备水溶性共聚取代聚炔p
‑
l
0.8
‑
co
‑
d
0.2
‑
ala
‑
nh2,具体如下:将p
‑
l
0.8
‑
co
‑
d
0.2
‑
ala
‑
nh2溶于二氯甲烷中,加入油溶性共聚物摩尔量1.5倍的三氟乙酸脱除boc保护基,在30℃反应24h。再加入碳酸钾除去三氟乙酸制得p
‑
l
0.8
‑
co
‑
d
0.2
‑
ala
‑
nh2。
[0084]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.8
‑
co
‑
d
0.2
‑
ala
‑
nh2,将p
‑
l
0.8
‑
co
‑
d
0.2
‑
ala
‑
nh2溶解于去离子水中,再加入与共聚物单体单元比为1:1的铜离子水溶液,30℃下反应24h后,获得负载铜离子的单手性螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.8
‑
co
‑
d
0.2
‑
ala
‑
nh2。
[0085]
结果:实施例3与实施例2结果基本相同。
[0086]
实施例4
[0087]
第一步制备油溶性共聚取代聚炔p
‑
l
0.7
‑
co
‑
d
0.3
‑
ala
‑
boc,具体如下:将n
‑
叔丁氧羰基
‑
l
‑
丙氨酸或n
‑
叔丁氧羰基
‑
d
‑
丙氨酸与氯甲酸异丁酯和炔丙胺进行酰胺化反应,得到取代炔单体m
‑
l(or d)
‑
ala
‑
boc;m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc以投料比为7:3在总单体摩尔量1%的(nbd)rh
b
‑
(c6h5)4的作用下30℃进行溶液聚合24h,再经过后处理得到油溶性共聚取代聚炔p
‑
l
0.7
‑
co
‑
d
0.3
‑
ala
‑
boc;
[0088]
第二步制备水溶性共聚取代聚炔p
‑
l
0.7
‑
co
‑
d
0.3
‑
ala
‑
nh2,具体如下:将p
‑
l
0.7
‑
co
‑
d
0.3
‑
ala
‑
nh2溶于二氯甲烷中,加入油溶性共聚物摩尔量1.5倍的三氟乙酸脱除boc保护基,在30℃反应24h。再加入碳酸钾除去三氟乙酸制得p
‑
l
0.7
‑
co
‑
d
0.3
‑
ala
‑
nh2。
[0089]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.7
‑
co
‑
d
0.3
‑
ala
‑
nh2,将p
‑
l
0.7
‑
co
‑
d
0.3
‑
ala
‑
nh2溶解于去离子水中,再加入与共聚物单体单元比为1:1的铜离子水溶液,30℃下反应24h后,获得负载铜离子的单手性螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.7
‑
co
‑
d
0.3
‑
ala
‑
nh2。
[0090]
结果:实施例4与实施例2结果基本相同。
[0091]
实施例5
[0092]
第一步制备油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc,具体如下:将n
‑
叔丁氧羰基
‑
l
‑
丙氨酸或n
‑
叔丁氧羰基
‑
d
‑
丙氨酸与氯甲酸异丁酯和炔丙胺进行酰胺化反应,得到取代炔单体m
‑
l(or d)
‑
ala
‑
boc;m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc以投料比为6:4在总单体摩尔量0.1%的(nbd)rh
b
‑
(c6h5)4的作用下30℃进行溶液聚合24h,再经过后处理得到油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc;
[0093]
第二步制备水溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体方法如实施例1。
[0094]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2具体方法如实施例1。
[0095]
结果:实施例5与实施例1结果相似,只是实施例5相较于实施例1,由于催化剂使用量从1%降低到0.1%,聚合物p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc的产率大幅下降(接近19.6%)。
[0096]
实施例6
[0097]
第一步制备油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc,具体如下:将n
‑
叔丁氧羰基
‑
l
‑
丙氨酸或n
‑
叔丁氧羰基
‑
d
‑
丙氨酸与氯甲酸异丁酯和炔丙胺进行酰胺化反应,得到取代炔单体m
‑
l(or d)
‑
ala
‑
boc;m
‑
l
‑
ala
‑
boc和m
‑
d
‑
ala
‑
boc以投料比为6:4在总单体摩尔量10%的(nbd)rh
b
‑
(c6h5)4的作用下30℃进行溶液聚合24h,再经过后处理得到油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc;
[0098]
第二步制备水溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体方法如实施例1。
[0099]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2具体方法如实施例1。
[0100]
结果:实施例6与实施例1结果相似,只是实施例5相较于实施例1,由于催化剂使用量增大,最终聚合物p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc的产率有所上升(接近100%),然而,催化剂使用量的增加致使聚合的成本大幅提高。
[0101]
实施例7
[0102]
第一步制备油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc,具体方法如实施例1。
[0103]
第二步制备水溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体如下:将p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc溶于三氯甲烷加入油溶性共聚物摩尔量1.5倍的三氟乙酸脱除boc保护基,在30℃反应24h。再加入碳酸钾除去三氟乙酸制得p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2。
[0104]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2具体方法如实施例1。
[0105]
结果:实施例7与实施例1结果基本相同。
[0106]
实施例8
[0107]
第一步制备油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc,具体方法如实施例1。
[0108]
第二步制备水溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体如下:将p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc溶于二氯甲烷中,加入油溶性共聚物摩尔量0.5倍的三氟乙酸脱除boc保护基,在30℃反应24h。再加入碳酸钾除去三氟乙酸制得p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2。
[0109]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2具体方法如实施例1。
[0110]
结果:实施例8与实施例1结果相似,只是实施例8相较于实施例1,产物(p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2)的boc基团脱除率有所降低,根据1hnmr谱图,从99.5%下降到了70.5%。
[0111]
实施例9
[0112]
第一步制备油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc,具体方法如实施例1。
[0113]
第二步制备水溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体如下:将p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc溶于二氯甲烷中,加入油溶性共聚物摩尔量2倍的三氟乙酸脱除boc保护基,在30℃反应24h。再加入碳酸钠、醋酸钾、醋酸钠、碳酸氢钾、碳酸氢钠、磷酸钠或磷酸钾除去三氟乙酸制得p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2。
[0114]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2具体方法如实施例1。
[0115]
结果:实施例9与实施例1结果相同。
[0116]
实施例10
[0117]
第一步制备油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc,具体方法如实施例1。
[0118]
第二步制备水溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体方法如实施例1。
[0119]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体如下:将p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2溶解于去离子水中,再加入与共聚物单体单元比为0.1:1的铜离子水溶液,30℃下反应24h后,获得负载铜离子的单手性螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2。
[0120]
结果:实施例10与实施例1结果相似,只是金属离子的含量不足以诱导整个聚合物主链形成螺旋结构,相比于实施例1中的负载铜离子的单手性螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2的光学活性明显降低60%。
[0121]
实施例11
[0122]
第一步制备油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc,具体方法如实施例1。
[0123]
第二步制备水溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体方法如实施例1。
[0124]
第三步制备负载铜离子的螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体如下:将p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2溶解于去离子水中,再加入与共聚物单体单元比为2:1的铜离子水溶液,30℃下反应24h后,获得负载铜离子的单手性螺旋共聚取代聚炔cu
2
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2。
[0125]
结果:实施例11与实施例1结果相似,只是金属离子造成浪费。
[0126]
实施例12
[0127]
第一步制备油溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
boc,具体方法如实施例1。
[0128]
第二步制备水溶性共聚取代聚炔p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体方法如实施例1。
[0129]
第三步制备ag
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2,具体如下:将p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2溶解于去离子水中,再加入与共聚物单体单元比为1:1的银离子水溶液,30℃下反应24h后,获得负载金属离子的单手性螺旋共聚取代聚炔ag
‑
p
‑
l
0.6
‑
co
‑
d
0.4
‑
ala
‑
nh2。
[0130]
图11为ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的cd和uv
‑
vis光谱图。不同投料比的ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的cd光谱在301nm和350nm处对应的表现出镜像的cd信号。且紫外吸收位置随着ee%值的减小350nm处的峰逐消失,cd信号强度也随着ee%值的减小而逐渐减小。具有ee%=80%的ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的cd信号强度大幅下降。之后掺入的异构体越多下降越快。这说明ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2络合物极不稳定。
[0131]
图12为ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的比旋光度曲线图。ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2与cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2在宏观状态方面也有很大的不同,cu
2
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2是十分澄清的状态,而ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2溶液中具有红黑色的沉淀,很可能是因为形成了大颗粒的氧化银或者是形成了多种配位络合物。所以测比旋光度的时候,是在络合2h之内完成的。这是因为2h之后,ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2络合溶液浑浊不透明,无法进行旋光测试。在室温下,ag
‑
p
‑
l
r
‑
co
‑
d1‑
r
‑
ala
‑
nh2的比旋光度在
‑
144
°
至 139
°
范围内,比旋光度与共聚物中单体单元的对映体过量百分比呈非线性关系,但是用ag
诱导过后的聚合物比旋光度相对于未络合之前聚合物比旋光度只增加了将近40多倍。说明这类络合物的“大多数规则”服从程度低。进一步说明金属离子种类影响着优异光学活性单手螺旋共聚取代聚炔的形成。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。