用于检测样本中的核酸的数位聚合酶连锁反应方法
1.相关申请
2.本技术根据美国专利法第119条(35 u.s.c.
§
119)主张于2018年10月9日提出申请的美国临时申请第62/743,149号的权益,其全部内容通过引用合并于本文。
技术领域
3.本发明涉及一种检测样本中核酸(nucleic acid,na)分子的方法。更具体而言,本发明涉及一种用于分析核酸片段的改良的基于数位pcr的方法。本发明可用于研究及诊断应用,具提高的灵敏度及准确性。本发明还提供一种试剂盒,用于执行本文所述的检测样本中核酸的方法。
背景技术:
4.已经针对许多研究及诊断应用开发了用于核酸检测的各种技术。数位聚合酶连锁反应(digital polymerase chain reaction,dpcr)被认为是分析基因复制数变异(copy number variations,cnvs)、基因表现、遗传突变及单核苷酸多型性(single
‑
nucleotide polymorphisms,snps)的最精密的定量方法之一。它的受欢迎程度不断提高。许多公司已经设计了公司特定的实验方法,硬件及软件应用程序(dong等人,2015年;morley,2014年;zhao等人,2016年)。
5.最普遍的模型是基于液滴以及基于滴定盘的数位pcr方法。目前基于液滴的数位pcr是由bio
‑
rad公司所贩卖。qx200 droplet digital pcr(ddpcr)仪是bio
‑
rad公司最近贩卖的产品,是结合微流控与界面活性剂化学技术将pcr反应分为油包水液滴以进行绝对核酸定量的最先进模型之一(hindson等人,2011年)。这种方法允许每次运行每个样本分析近20,000纳升大小的液滴,使其成为同类仪器中最有效的方法。另一方面,以qiagen公司(http://www.captodayonline.com/high
‑
throughput
‑
digital
‑
pcr
‑
system
‑
1017/)为例,基于滴定盘的数位pcr在滴定盘上进行反应。对于这种方法,将dna分子稀释并分配到例如96孔滴定盘中,以进行独立的pcr反应。扩增后,通过基因特异性序列探测并定量所有的孔,以鉴定具有阳性反应的孔。
6.作为基于pcr的核酸检测技术,数位pcr通常需要成对的引子进行扩增,并需要使用探针来检测目标核酸,将样本稀释并分配,以便分离其中的核酸片段以进行独立的反应,每个独立的反应具有非常有限数量的目标核酸分子。如此一来,可以在每个分区内分别进行pcr反应,并确定每个分区中的信号为阴性或阳性,因此,可以基于泊松分布(poisson distribution),通过对正向分区(检测到序列)与负向分区(未检测到序列)的数量进行计数,以确定原始样本中核酸序列复制的确切数量。通常,数位pcr的实验过程包括以下几个步骤:1)稀释目标dna分子;2)将充分分离的目标dna片段分为离散的液滴/腔室,每个液滴/腔室还包含其他用于扩增及信号检测所需的成分;3)使用针对目标基因“内部(internal)”区域的“成对(paired)”基因特异性引子进行pcr扩增;4)使用荧光探针检测荧光信号;5)基于泊松分布对阳性与阴性反应进行定量;以及6)交叉样本比较以确定显著性。
7.无细胞核酸样本为易于获得、非侵入性的遗传材料,在诊断多种疾病中越来越受欢迎(wagner,2012年)。这些遗传物质从体内所有细胞释放,包括正常细胞、患病细胞以及微生物。理论上,无细胞核酸可能存在于多种体液中,包括血液、唾液、尿液、白带、精液、淋巴液以及汗液(chiu与yu,2019年;nai等人,2017年;wagner,2012年)。尽管具有许多优点,但无细胞核酸样本的数量通常很少,而造成这些样本难以操作,因此容易在实验过程中丧失。此外,无细胞核酸也会高度断裂。这些都特别显示出珍贵材料的严重问题。
8.现行的数位pcr在循环无细胞核酸分析方面仍然面临许多挑战。一些方法被提出来改善当前的数位pcr,例如适当保存核酸样本以减少无细胞dna(cell
‑
free dnas,cfdnas)的降解,有效的纯化例如硅胶膜收集足够量的cfdnas进行检测,并通过高灵敏度毛细管电泳提高检测灵敏度。现行的数位pcr还具有缺点,即基于特定的已知突变位点设计探针以检测核酸中的某些突变,但其不能涵盖所有遗传变异,因此限制了对其他潜在变异或突变的测试。
9.因此,需要开发用于核酸检测的改良的数位pcr方法。
技术实现要素:
10.本发明提供一种用于样本中核酸检测的改良的数位pcr,称为数位t
‑
寡引子pcr(数位top
‑
pcr)。本发明的数位top
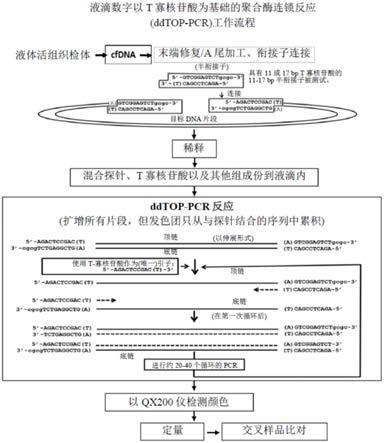
‑
pcr的特征在于使用与样本中所有核酸片段的末端连接的均质衔接子(homogeneous adapter,ha)以及识别ha中互补序列的单一型引子(t/u寡核苷酸),并作为正向及反向引子以进行扩增。本发明的数位top
‑
pcr可以扩增样本中的所有核酸片段,进而可藉由增加的灵敏度,特别是降低伪阴性率,以实现目标核酸的后续检测。
11.具体而言,本发明提供一种用于分析样本中的核酸的方法,该样本包含一或多个线性、双链核酸片段(na片段),该方法包括以下步骤:
12.(a)使该样本进行3'
‑
a尾加工反应,允许在该na片段的3'
‑
尾添加腺嘌呤核苷酸(a)以产生3'
‑
腺嘌呤核苷酸(3'
‑
a)突出端核酸片段(3'
‑
a突出的na片段);
13.(b)提供3'
‑
胸腺嘧啶(t)或3'
‑
尿嘧啶(u)核苷酸突出的双链均质衔接子,其包含携带5'
‑
磷酸的p寡核苷酸链以连接至该3'
‑
a突出的na片段以及带有3'
‑
t或3'
‑
u而不带有5'
‑
磷酸的一t/u寡核苷酸链,其中该t/u寡核苷酸链与该p寡核苷酸链互补,除了在该t/u寡核苷酸链的3'
‑
t或3'
‑
u之外;
14.(c)使(a)的样本进行连接反应,以使该均质衔接子在两端与该3'
‑
a突出的na片段连接,以产生衔接子连接的核酸片段(衔接子连接的na片段);
15.(d)将(c)的样本与聚合酶连锁反应(pcr)试剂及检测试剂结合以提供准备好扩增/检测的样本,其中该pcr试剂包括用于扩增的单一型引子,其具有该t/u寡核苷酸链的核酸序列,且该检测试剂包括一或多种用于检测的荧光探针,其产生荧光信号并与该na片段特异性杂交;
16.(e)将(d)的该准备好扩增/检测的样本分成多个分区,每个分区包含该衔接子连接的na片段的有限个复制;
17.(f)以该衔接子连接的na片段为模板,以该单一型引子作为正向及反向引子在每个分区中进行pcr,以扩增该衔接子连接的na片段;以及
18.(g)评估每个部分的该荧光信号。
19.于一些具体实施例中,步骤(g)包括基于荧光信号的强度确定液滴/级分为正或负,且随后计算具有一正信号的液滴/级分的总数(计数)。
20.于一些具体实施例中,在步骤(e)中,超过50%的分区包含不超过一个复制的该衔接子连接的na片段。
21.于一些具体实施例中,在步骤(e)中,每个分区包含至少一个复制的该衔接子连接的na片段。
22.于一些具体实施例中,该na片段包含可指示该个体的健康/患病状态的核酸序列。
23.于一些具体实施例中,步骤(b)的该均质衔接子不自体连接。
24.于一些具体实施例中,步骤(b)的该均质衔接子在其t/u寡核苷酸链中具有3'
‑
t或3'u突出端,且在其p寡核苷酸链中具有3'
‑
非a突出端。
25.于一些具体实施例中,步骤(b)的该均质衔接子的一端为3'
‑
t突出端,另一端为钝端。
26.于一些具体实施例中,该样本获自体液,包括,但不限于,血液、尿液、唾液、眼泪、汗液、母乳、鼻分泌物、羊水、精液,以及阴道液。
27.于一些具体实施例中,该样本中的该na片段为无细胞dna(cell
‑
free dnas,cfdnas)。
28.于一些具体实施例中,在步骤(a)之前,本文所述的方法还包括对该na片段进行末端修复反应。
29.于一些具体实施例中,步骤(f)的该pcr通过油乳剂或液滴pcr或基于孔的pcr进行。
30.于一些具体实施例中,步骤(g)的测定通过使用荧光探针的流式细胞分析法进行。
31.于一些具体实施例中,本发明的方法包括步骤(a)至(g)以及可任选的步骤(a)':如果该na片段包含线性、单链rnas,在步骤(a)之前对该样本进行反转录pcr(reverse transcription
‑
pcr,rt
‑
pcr),以将该rnas转换为线性、双链互补dna(互补dna,cdna)。
32.本发明还提供一种用于执行如本文所述的检测样本中的核酸片段的方法的试剂盒。特定而言,该试剂盒包括
33.(i)衔接子连接试剂,包括均质衔接子、连接缓冲液,以及连接酶,其中该均质衔接子包含带有5'
‑
磷酸的p寡核苷酸链以及带有3'
‑
t或3
‑
u而不带有5'
‑
磷酸的t/u寡核苷酸链,其中该t/u寡核苷酸链与该p寡核苷酸链互补,除了在该t/u寡核苷酸链的3'
‑
t或3'
‑
u处,其中该均质衔接子能够在两端与该核酸片段连接,其中该核酸片段具有3'
‑
a突出端;
34.(ii)pcr试剂,包含具有该t/u寡核苷酸链的核酸序列的单一型引子(唯一的引子)、dntps、pcr缓冲液,以及dna聚合酶;以及
35.(iii)检测试剂,包含一或多种可检测探针,其具有与该核酸片段特异性杂交的互补序列。
36.于一些具体实施例中,该试剂盒进一步包括使用说明书,其中该使用说明书包括用于执行包括本文所述的步骤(a)至(g)的方法的说明书。
37.在以下的描述中阐述了本发明的一或多个具体实施例的细节。通过以下几个具体实施例的详细描述以及所附申请专利范围,本发明的其他特征或优点将变得显而易见。
附图说明
38.当结合附图阅读时,将更好地理解前述发明内容以及以下对本发明的详细描述。为了说明本发明,在附图中示出了目前较佳的具体实施例。然而,应当理解的是,本发明不限于所示的精确布置及手段。
39.图1所示为本发明的方法的程序。p寡核苷酸:5
’‑
gtcggagtctgcgc
‑3’
(seq id no:24)。t
‑
寡核苷酸:5
′‑
agactccgac(t)
‑3′
(seq id no:23)。
40.图2所示为本发明的方法与传统的基于pcr的检测方法之间的差异。cfdna样本包含一组cfdna片段,这些片段带有来自基因组来源的随机断裂。在传统的基于pcr的检测方法中,仅覆盖第一给定引子结合位点与第二给定引子结合位点的cfdnas片段“g”可被扩增及检测。结果,检测的灵敏度受到限制,特别是当核酸含量低时,灵敏度甚至会更差。相反地,于本发明的方法中,所有cfdna片段皆可被均匀且普遍地扩增,且在这种扩增之后,不仅片段“g”而且还有其他片段“a”至“f”(仅具有一个引子结合位点,或甚至没有任何引子结合位点)被扩增,可以检测到所有皆来自相同目标致病核酸(相同基因组来源),例如,使用一或多种带有可检测标记的探针,该标记能够与目标病原性核酸内的任何区域特异性杂交;结果,由于检测前的扩增同样(非特异性地)适用于所有核酸片段,因此提高了检测的灵敏度并且可以最小化伪阴性,因此每个扩增的核酸片段的相对量可以代表原始样本中存在的相对量。
41.图3所示为使用包含不同含量的部分及完整模板的样本,从传统ddpcr以及ddtop
‑
pcr方法产生的阳性液滴的计数。泳道1与2(a05与a06对照)为分别使用t/u寡核苷酸引子以及n
‑
myc基因特异性引子从5'引子结合位点缺失的nagk基因序列所扩增而产生,其余部分(泳道3
‑
16或b05
–
h06)是由混合模板所产生的,该模板包含不同含量(100%
‑
0%)的部分(标记为“h”,或5'引子结合位点缺失)以及完整(标记为“f”,或带有两个引子结合位点)的模板。所有样本均使用相同的n
‑
myc探针进行检测。ddtop
‑
pcr的计数在字母后标有“05”,而其对应的ddpcr的计数则并排标有“06”。图中的数字代表计数(复制数/微升)。每个样本总共20微升,用于qx200ddpcr仪计数。ch1,通道1,由qx200定义。
42.图4所示为ddpcr与ddtop
‑
pcr之间荧光信号强度的比较。与图3相同,泳道1与2(a05与a06对照)为分别使用t/u寡核苷酸引子以及n
‑
myc引子从5'引子结合位点缺失的nagk模板所扩增而产生,其余部分(泳道3
‑
16或b05
–
h06)是由混合模板所产生的,该模板包含不同含量(100%
‑
0%)的部分(标记为“h”,或5'引子结合位点缺失)以及完整(标记为“f”,或带有两个引子结合位点)的模板。所有样本均使用相同的n
‑
myc寡核苷酸探针进行检测。请注意,显示为黑点的液滴不被视为正液滴,因为其强度低于预设的阈值。
43.图5所示为表1中列出的序列。t寡核苷酸:5'
‑
agc gct aga ctc cga ct
‑
3'(seq id no:1)。p寡聚核苷酸,5'
‑
gt cgg agt cta gcg ct
‑
3'(seq id no:2)。
具体实施方式
44.除非另有定义,否则本文使用的所有技术及科学术语具有与本发明所属领域的技术人员通常所理解的相同含义。
45.如本文所用,冠词“一”以及“一个”是指冠词的语法对象中的一个或多个(即,至少一个)。举例而言,“一个组件”是指一个组件或一个以上组件。
[0046]“包括(动词)”或“包括(动名词)”等词通常以包含/包含的意义使用,其代表允许存在一种或多种特征、成分或组成分。“包括(动词)”或“包括(动名词)”等词涵盖“组成”或“由...组成”等词。
[0047]
如本文所用,“大致”、“大约”或“大概”通常可以指给定值或范围的20%以内,特别是10%以内,更特别是5%以内。在此给出的数值为近似的,表示如果没有明确指出,则可以推断出“大致”、“大约”或“大概”等词。
[0048]“多核苷酸”或“核酸”等词是指由核苷酸单元组成的聚合物。多核苷酸包括天然存在的核酸,例如脱氧核糖核酸(deoxyribonucleic acid,“dna”)以及核糖核酸(ribonucleic acid,“rna”)以及核酸类似物,包括具有非天然存在的核苷酸的核酸类似物。可以使用例如自动dna合成仪合成多核苷酸。“核酸”一词通常是指大的多核苷酸。将理解的是,当核苷酸序列由一dna序列(即,a、t、g、c)表示时,其还包括其中以“u”替代“t”的rna序列(即,a、u、g、c)。“寡核苷酸”一词指相对短的核酸片段,典型地小于或等于150个核苷酸长度,例如,5至150之间。寡核苷酸可以根据需要设计并合成。就引子而言,其长度通常为5至50个核苷酸,特别是8至30个核苷酸。就探针而言,其长度通常为10至100个核苷酸,特别是15至30个核苷酸。
[0049]
如本文所用,“互补的”一词是指两个多核苷酸的相互作用表面的拓扑兼容性或匹配在一起。因此,这两个分子可被描述为互补的,此外,接触表面特性彼此互补。如果第一多核苷酸的核苷酸序列与第二多核苷酸的多核苷酸结合伴侣的核苷酸序列相同,则第一多核苷酸与第二多核苷酸互补。因此,序列5'
‑
tatac
‑
3'的多核苷酸与序列5'
‑
gtata
‑
3'的多核苷酸互补。
[0050]
如本文所用,目标核酸可以指在一样本中检测到的特定目标核酸。目标核酸包括但不限于dna,例如基因组dna、线粒体dna、cdna及其类似物,以及rna,例如mrna、mirna及其类似物。目标核酸可源自任何来源,包含天然来源或合成来源。例如,目标核酸可来自动物或病原体来源,包含,但不限于,哺乳动物如人类,以及病原体如细菌、病毒以及真菌。目标核酸可从任何体液或组织(例如,血液、尿液、皮肤、头发、粪便,以及黏液)或环境样本(例如,水样本或食物样本)获得。于一些具体实施例中,目标核酸可为相同来源(例如,来自正常或患病个体或病原体的相同基因)的核酸分子的集合,但是具有各种长度。例如,编码b型肝炎表面抗原(hepatitis b surface antigen,hbsag)的基因的许多片段可作为存在于测试样本中的各种长度的“目标”核酸片段。由于每个目标核酸分子均包含至少一部分hbsag基因,因此具有与hbsag基因内各个位置对应(或互补)的序列的探针或引子可用于检测目标核酸片段。再例如,目标核酸可为含有遗传突变的核酸(例如,指示疾病如癌症的单核苷酸多型性(single nucleotide polymorphism,snp)。
[0051]
如本文所用,“引子”一词是指可用于扩增方法,例如,聚合酶连锁反应(pcr),中以扩增目标核苷酸序列的寡核苷酸。在传统pcr中,需要至少一对引子,包括一个正向引子和一个反向引子来进行扩增。通常,对于由一( )链以及一(
‑
)链组成的要扩增的目标dna序列,正向引子为一种寡核苷酸,可与该(
‑
)链的3'端杂交,进而在反应条件下引发新的( )链的聚合;而反向引子为另一种寡核苷酸,可在反应条件下与该( )链的3'端杂交并因此可以在该反应条件下引发新的(
‑
)链聚合。特定而言,例如,正向引子可具有与( )链的5'端相同的序列,而反向引子可具有与(
‑
)链的5'端相同的序列。通常,用于扩增目标核酸序列的正
向引子以及反向引子的序列不同于彼此。如本文所用,一“单个”引子是指仅一种类型的引子,所有引子均具有相同的序列,而非一对具有不同序列的引子,一个为正向引子,另一个为反向引子。
[0052]
如本文所用,“杂交”一词应包括核酸链通过碱基配对与互补链结合的任何过程。相关方法为本领域所熟知的,且描述于,例如,sambrook等人,molecular cloning:a laboratory manual,第二版,冷泉港实验室出版社(1989年),以及frederick ma等人,current protocols in molecular biology,john wiley&sons公司(2001年)。通常,严格条件应选择为在规定的离子强度以及ph值下,比指定序列的热熔点(t
m
)低约5至30℃。更典型地,在规定的离子强度以及ph值下,严格条件被选择为比指定序列的t
m
低约5至15℃。例如,严格的杂交条件将是其中盐浓度小于约1.0m钠(或其他盐类)离子,通常为约0.01至约1m钠离子浓度,其在约ph 7.0至约ph 8.3下,且温度为针对短探针(例如,10至50个核苷酸)至少约25℃,对于长探针(例如,大于50个核苷酸)至少约55℃。长探针(例如,大于50个核苷酸)的示例性非严格或低严格条件将包含20mm tris,ph 8.5、50mm kcl,以及2mm mgcl2的缓冲液,反应温度为25℃。
[0053]
如本文所用,“突出端”是指在线性双链核酸分子的末端的单个未配对核苷酸的片段或更长的未配对核苷酸的片段。未配对的核苷酸可以在3'或5'端,分别产生3'或5'突出端。“3'
‑
a突出端”是指未配对的核苷酸存在于3’端,并且由一或多个腺嘌呤(a)核苷酸组成。“3'
‑
非a突出端”是指未配对的核苷酸存在于3'端,并且不包括任何腺嘌呤(a)核苷酸。“3
‑
t突出端”是指未配对的核苷酸存在于3’端,并且由一或多个胸腺嘧啶(t)核苷酸组成。
[0054]“单一(single)”、“同质(homogenous)”或“通用(universal)”引子是指在pcr反应中仅存在一种具有相同序列的引子,而非一对引子。“异质引子(heterogenous primers)”一词是指在pcr反应中存在至少一对配对的引子,每个引子具有不同于彼此的序列。
[0055]
如本文所用,“衔接子”一词是指可连接至双链核酸分子末端的寡核苷酸。衔接子的长度可为10至50个碱基,较佳为10至30个碱基,更佳为10至20个碱基。少于10个核苷酸的长度可能会降低退火的特异性。长度超过20个核苷酸可能并不划算。一“均质(homogenous)”衔接子一词是指用于连接至双链核酸分子的两端的单一类型的衔接子。“异质(heterogenous)”衔接子一词是指至少两种类型的衔接子,它们具有不同于彼此的核苷酸序列,一种用于连接至该双链核酸分子的5'端,另一种用于连接至该双链核酸分子的3'端。
[0056]
为了扩增样本中的核酸片段,我们已经开发了一种以t寡核苷酸为基础的聚合酶连锁反应(top
‑
pcr)技术,该方法是使用由p寡核苷酸与t寡核苷酸所组成并连接到所有核酸片段末端的均质衔接子,然后将t寡核苷酸作为单一引子,以无差别地扩增样本中的所有核酸片段。参见美国专利第10,407,720号,其全部内容通过引用并入本文。
[0057]
于本发明中,发现通过使用top
‑
pcr技术可显著改善传统的数位pcr。通过使用top
‑
pcr进行扩增,以数位方式执行本发明的方法以进行核酸检测,以使反应中的所有核酸均等比例地扩增,并且可以通过一或多个序列特异性探针以更高的灵敏度检测目标核酸。如以下提供的实施例所示,相较于使用配对pcr引子进行扩增的传统数位pcr,本发明的方法显示出增加至少约14%的灵敏度。
[0058]
图1为显示本发明的方法的过程的图。
[0059]
核酸样本
[0060]
dna样本(例如,cfdna样本)可获自含有待检测的特定目标核酸的任何样本,例如,体液或组织,例如,血液、尿液、皮肤、头发、粪便,以及黏液,或环境样本,例如,水样本或食物样本。可通过传统程序(例如,苯酚
‑
氯仿萃取或qiagen公司试剂盒)对样本进行处理,以从中分离并纯化dna。本发明的方法可用于dna及rna目标。针对dna样本,dna聚合酶可直接用于扩增。针对rna样本,首先需要使用反转录酶进行反转录步骤。
[0061]
末端修复以及a尾加工
[0062]
样本中的dna片段经过末端修复,在每个3’端都加上一个“a”,以提供3'a突出的dna片段。可使用传统方法或试剂盒执行末端修复以及a尾加工步骤,例如ultra end repair/da
‑
tailing module(neb公司,e7442s/l)。
[0063]
用于t/u寡引子pcr(top
‑
pcr)扩增的均质衔接子
[0064]
设计均质衔接子用于top
‑
pcr扩增。在均质衔接子中,一条链称为t/u寡核苷酸,在3'端具有一个额外的胸腺嘧啶或尿嘧啶核苷酸(t/u);另一条链称为p寡核苷酸,在5'端具有磷酸基,其3'端核苷酸没有多余的t或u。该衔接子可为钝黏性的(即一端是钝的,另一端是黏性的)或双黏性(即两端都是黏性的)衔接子。于一些具体实施例中,针对“钝黏性”衔接子,p寡核苷酸短一个碱基,并且与t/u寡核苷酸互补,除了在t/u寡核苷酸的3'端t/u处。于一些具体实施例中,针对“双黏性”衔接子,p寡核苷酸长于t/u寡核苷酸。本文使用的均质衔接子需要(i)t/u寡核苷酸具有额外的3'
‑
t/u(即,3'端的“t”或“u”突出端)并且没有5'
‑
磷酸盐;(ii)p寡核苷酸需要5'
‑
磷酸;以及(iii)t/u寡核苷酸与p寡核苷酸互补,但除了t/u寡核苷酸的3'
‑
t/u突出端。t/u寡核苷酸以及p寡核苷酸的长度及序列可能有所不同。于一些实施例中,p寡核苷酸序列为5
’‑
gtcggagtctgcgc
‑3’
(seq id no:24),且t/u寡核苷酸序列为5
’‑
agactccgac(t)
‑3’
(seq id no:23)。于一些实施例中,p寡核苷酸序列为5'
‑
gt cgg agt cta gcg ct
‑
3'(seq id no:2),且t/u寡核苷酸序列为5'
‑
agc gct aga ctc cga ct
‑
3'(seq id no:1)。此外,于一些具体实施例中,可使用“3'
‑
u”而非“3'
‑
t”,进而可以在扩增后通过使用“用户酶(user enzyme)”(尿嘧啶特异性切除试剂(uracil
‑
specific excision reagent),neb公司)将双链半衔接子(half adapter,ha)完全修剪掉。
[0065]
均质衔接子与dna片段的连接
[0066]
在末端修复以及a
‑
尾加工步骤之后,将均质衔接子连接至3'a
‑
突出dna片段的两个末端,以产生衔接子连接的dna片段,其中该衔接子的t/u寡核苷酸的3'
‑
t/u与3'
‑
a突出dna片段的3
‑
'a突出互补。可以在适当的条件下,例如,在热循环仪中约25℃整夜,在含有衔接子、3
′‑
a突出dna片段、连接酶,以及连接缓冲液的适当的连接混合物中进行连接。连接混合物可以直接进行pcr扩增,可以进行或不进行dna纯化。
[0067]
pcr/检测试剂
[0068]
连接后,将样本与pcr/检测试剂合并以提供可扩增/检测的样本。pcr试剂通常包括引子、核苷酸、聚合酶,以及缓冲液。检测试剂通常包括一或多种可检测探针。这些输入试剂可以单独添加到样本中的单独试剂的形式提供,或者可以将某些或全部试剂作为以预混合形式添加到样本中的试剂混合物来提供。pcr试剂通常包括选择用于促进扩增反应的缓冲液。镁离子,例如mgcl2有用地包含在缓冲液中。pcr试剂还包括核苷酸。通常以等摩尔浓度提供四种dntps(datp、dctp、dgtp,以及dttp)。各种pcr聚合酶可用于同一扩增中。合适的
聚合酶通常将在约75℃下具有最佳活性,并且在长时间作用后(例如,在高于95℃的温度下)保持该活性的能力。有用的聚合酶可包括,例如,taq dna聚合酶,例如dna聚合酶,例如的stoffel片段等。特定而言,pcr试剂包括具有如本文所述的t/u寡核苷酸链的核酸序列的单一型引子,作为用于扩增的正向与反向引子。可以将包括对目标核酸具有特异性的探针的检测试剂添加到样本中,并且可以检测到可检测的信号,例如由探针降解引起的荧光信号。
[0069]
区份(fractionation)/稀释(dilution)
[0070]
准备好扩增/检测的样本被分成多个分区,每个分区包含有限数目复制的衔接子连接的na片段。特定而言,大多数分区可能不包含任何复制,其他分区仅包含一个复制,其他分区可能包含两个复制,三个复制,甚至更多数量的复制。每个分区的复制可以根据需要进行调整。于一些具体实施例中,进行区份的程度使得超过50%的分区包含不超过一个复制的衔接子连接的na片段。于一些具体实施例中,进行区份的程度为每个分区包含至少一个复制的衔接子连接的na片段,例如每个分区1
‑
5个复制。区份可以如本领域已知的方式,在乳液液滴中或在多孔中进行,例如,lodrini等人,2017年,以及美国专利申请公开号2009/0053719以及20150099644中所述,其通过引用并入本文。于一些具体实施例中,通过水油乳化技术将样本分成油滴中的多个小反应。油滴使用液滴发生器产生。通常,从每20微升样本中大约形成20,000个油滴。
[0071]
t/u寡核苷酸为基础的pcr扩增
[0072]
区份后,使用游离t/u寡核苷酸作为唯一的pcr引子进行pcr扩增。如本文所用,游离t/u寡核苷酸是具有如本文所述的t/u寡核苷酸链的核酸序列的单个引子。游离t/u寡核苷酸是指未在衔接子中与其互补的p寡核苷酸形成的t/u寡核苷酸。以此方式,在两端与衔接子连接的所有dna片段均等比例扩增。
[0073]
检测/定量
[0074]
目标dna的检测可以通过本领域已知的许多方法进行,例如使用荧光探针的流式细胞分析法。
[0075]
于某些具体实施例中,具有可检测标记的探针例如荧光团(例如,fam、6
‑
荧光素亚酰胺)被用于检测。荧光探针具有与目标核酸片段特异性杂交的互补序列,其中荧光团在该目标核酸片段以pcr扩增时从该探针释放出来(产生阳性信号,表示检测到序列),而如果不存在目标核酸片段或没有目标核酸片段被扩增,则荧光团不从探针释放出来(生成阴性信号,表示未检测到序列)。于一些具体实施例中,pcr混合物中存在可检测的探针。
[0076]
传统的数位pcr使用通常基于目标核酸内部区域中的一特定位点(例如,突变位置)设计的探针,该目标核酸使用配对引子扩增;这样的探针不能涵盖所有的遗传变异,因此限制了在核酸中其它潜在生物标记的检测。相较之下,本发明的方法允许扩增样本中的所有核酸片段,因此可以使用能够与目标核酸内的任何区域特异性杂交的猎枪探针,并可通过提高灵敏度来实现检测目标核酸。请参阅图2的一特定具体实施例。根据本发明,不仅片段“g”(覆盖两个引子结合位点),而且还有其他片段“a”至“f”(仅具有一个引子结合位点或甚至没有任何引子结合位点),可以侦测全部源自相同目标病原菌的核酸,例如使用带有可侦测标记、能够与在该目标dna内的任何区域特异性杂交的猎枪探针,进而增加检测灵敏度以及使伪阴性降至最低。
[0077]
在检测之后,对阳性分区(检测到序列)相对于阴性分区(未检测到序列)的数量进行计数,以确定样本中目标核酸片段的预估量。可以根据本领域已知的方法进行定量,例如,如lodrini等人,2017年所述。于某些具体实施例中,可以在qx200ddpcr液滴读取仪中测量pcr扩增后的液滴,并使用quantasoft分析软件分析目标复制数。
[0078]
还提供用于执行如本文所述的检测样本中的核酸片段的方法的试剂盒。具体而言,该试剂盒包括
[0079]
(i)衔接子连接试剂,包括均质衔接子、连接缓冲液,以及连接酶,其中该均质衔接子包含带有5'
‑
磷酸的p寡核苷酸链以及带有3'
‑
t或3
‑
u而不带有5'
‑
磷酸的t/u寡核苷酸链,其中该t/u寡核苷酸链与该p寡核苷酸链互补,除了在该t/u寡核苷酸链的3'
‑
t或3'
‑
u处,其中该均质衔接子能够在两端与该核酸片段连接,其中该核酸片段具有3'
‑
a突出端;
[0080]
(ii)pcr试剂,包括具有该t/u寡核苷酸链的核酸序列的单一型引子,dntp(datp、dctp、dgtp,以及dttp)、pcr缓冲液,以及dna聚合酶;以及
[0081]
(iii)检测试剂,包含一或多种具有与该核酸片段特异性杂交的互补序列的可检测探针。
[0082]
于一些具体实施例中,该试剂盒进一步包含使用说明书。特定而言,该使用说明书包括用于执行本发明的方法的说明,包括步骤(a)至(g)。
[0083]
本发明的实用性及优点
[0084]
本发明的方法可用于诊断或预后,特别是在基于cfdna的检测中。已经描述了检测包含cfdna的体液样本的非侵入性方法,其可用于诊断基因缺陷、传染源以及疾病,特别是对于早期检测以及至少预后具有价值,因为即使患病的组织,例如肿瘤,被去除后,仍可获得cfdna。但是,传统的pcr,包括qpcr或dpcr,被设计为依赖模板,需要至少一对引子,因此不适合cfdna检测,因为cfdna作为模板的质量与数量通常不高,因此灵敏度有限,如果pcr循环数增加,则可能会发生偏差。本发明的方法,通过使用由p寡核苷酸与t/u寡核苷酸所组成的均质衔接子,连接至dna,以及该t/u寡核苷酸作为单一引子,能够以任何初始数量等比例扩增一样本中的所有dna,并可以使用特定探针检测目标dna,以提高灵敏度,而不会产生重大偏差(伪阴性)。
[0085]
通过以下实施例进一步说明本发明,提供这些实施例是为了说明而非限制。根据本公开,本领域技术人员应当理解的是,可在所公开的特定具体实施例中进行许多改变,并且在不脱离本发明的精神与范围的情况下仍可获得类似或相似的结果。
[0086]
实施例
[0087]
尽管数位pcr(dpcr)是分析遗传突变与复制数变异(copy number variations,cnvs)的有力方法,但由于cfdna片段的数量少且高度断裂,这可能会造成使用双引子扩增的传统数位pcr方法产生伪阴性,因此不适合用于临床样本中的无细胞dna(cfdnas)分析。为解决该问题,我们开发了液滴数位t/u寡聚引子聚合酶连锁反应(droplet digital t/u oligo
‑
primed polymerase chain reaction,ddtop
‑
pcr),该反应有赖于衔接子、无差异性地扩增所有cfdna片段,然后使用标记fam或hex的寡聚探针进行检测,fam或hex在延伸过程中从特定目标生成发色团。随后通过qx200 ddpcr仪检测荧光信号。结果显示,ddtop
‑
pcr能够检测5'引子结合位点缺失的n
‑
myc序列,而ddpcr不能。以含有5'引子结合位点缺失的构筑体及/或双重引子结合位点完整的n
‑
myc构筑体的片段的样本进行的进一步测试显示,
ddtop
‑
pcr相较于传统ddpcr的灵敏度提高了约14%,尽管信号强度有所降低。这些概念验证实验证明了在液体活组织检体中分析cfdna方面,ddtop
‑
pcr优于其相对的方法。
[0088]
1.材料与方法
[0089]
1.1.选殖n
‑
myc序列作为分析标准
[0090]
为了优化实验条件以及为分析复制数变异提供标准,基于由ncbi数据库检索的人类第2号染色体grch38.p12初级组合(登录号:nc_000002.12),使用针对人类n
‑
myc序列的引子,以构筑带有n
‑
myc基因序列的载体。使用正向引子5'
‑
aag ggg tgc tct cca att ct
‑
3'(seq id no:13)以及反向引子5
’‑
cgg ttt agc cac caa ctt tc
‑3’
(seq id no:14)由be2c细胞株的基因组dna中选殖/扩增n
‑
myc扩增子(157bp)。使用正向引子5'
‑
ccc ctt tcc cgc tat atc tt
‑
3'(seq id no:15)以及反向引子5
’‑
atg cag ggt ttg atg gga ta
‑3’
(seq id no:16)由be2c细胞株的基因组dna中选殖/扩增nagk扩增子(172bp)。使用q5 high
‑
fidelity 2x master mix(neb公司,麻州,美国)进行聚合酶连锁反应(pcr),以扩增目标扩增子。在含有相应引子对、be2c基因组dna(10ng)、1x q5 high
‑
fidelity master mix的混合物(50μl)中进行pcr反应。然后将混合物于98℃作用1分钟,然后于98℃作用20秒,60℃作用30秒,以及72℃作用10秒,共30个循环。混合物随后在72℃下作用2分钟以进行最终延伸。然后分析pcr产物,并按照说明手册,使用qiaquick凝胶萃取试剂盒(qiagen公司,nw,德国)从2%琼脂糖凝胶中萃取具有预期大小的扩增子,然后进行凝胶dna萃取。
[0091]
1.2将半衔接子(half
‑
adapter,ha)连接到n
‑
myc以及nagk标准模板
[0092]
首先通过退火16个单体单元的p寡核苷酸(5
’‑
pgtcggagtctagcgct
‑
3c6
‑3’
)(seq id no:2)以及17单体单元t/u寡核苷酸(5
’‑
amc6
‑
agcgctagactccgact
‑3’
)(seq id no:1),以1:1的摩尔比在95℃下作用5分钟,然后使用热循环仪将温度逐渐降低至4℃,制备半衔接子(ha)。
[0093]
连接前,以用于illumina定序的ultra
tm ii dna库制备试剂盒(neb公司,麻州,美国)稍微修改,首先对10ng的157bp n
‑
myc扩增子以及172bp nagk扩增子进行末端修复以及3'a尾加工。然后,使用例如ha与扩增子的比例为50:1进行连接,并将反应在热循环仪中于16℃作用整夜。
[0094]
需要时,无需纯化即可对连接混合物进行直接top
‑
pcr。反应在一混合物(50μl)中进行,该混合物使用t/u寡核苷酸(5'
‑
agcgctagactccgact
‑
3')(seq id no:1)1μm,连接混合物(5μ),1x phusion hf反应缓冲液(thermo fisher scientific公司,麻州,美国),该缓冲液包含mg
2
(1.5mm)、phusion高保真热启动dna聚合酶(1u)以及dntps(1mm)。然后该混合物于98℃下作用1分钟,然后于98℃作用20秒,57℃作用30秒,以及72℃针对剪切的gdna作用1分钟、针对标准模板作用10秒,共30个循环。混合物随后在72℃下作用5分钟以进行最终延伸。使用qiaquick pcr纯化试剂盒(qiagen公司,nw,德国)根据说明书指示纯化top
‑
pcr扩增的n
‑
myc与nagk,亦即分别为ha
‑
n
‑
myc
‑
ha以及ha
‑
nagk
‑
ha,并使用qubit dsdna hs分析试剂盒(thermo fisher scientific公司,麻州,美国)进行定量。
[0095]
1.3标准模板的构筑
[0096]
为了测试t/u寡核苷酸与探针特异性。制备phe
‑
n
‑
myc
‑
ha与phe
‑
nagk
‑
ha模板的不同比例组合。制备10倍序列稀释的phe
‑
n
‑
myc
‑
ha与phe
‑
nagk
‑
ha模板的比例为100:1至1:100。以稍微修改的方法制备ddpcr混合物。在液滴产生之前,ddpcr反应(20μl)包括t/u寡核
苷酸引子(8μm)、mpb1 2(0.25μm)、npb2(0.25μm)、dna模板(4.0μl)、1
×
ddpcr
tm
探针超混合物(无dutp)(bio
‑
rad公司,加州,美国)。制备ddpcr液滴混合物,并如上所述进行ddpcr反应。
[0097]
1.4gdna
‑
ha的测试
[0098]
使用ddtop
‑
pcr进行gdna混合物中n
‑
myc与nagk目标的特异性扩增。使用上述ddpcr参数将构筑的gdna
‑
ha模板用于本实验。本实验使用10倍序列稀释的100ng至100pg范围内的不同输入量的剪切gdna
‑
ha。在液滴产生之前,ddpcr反应(20μl)包含t/u寡核苷酸引子(8μm)、mpb1 2(0.25μm)、npb2(0.25μm)、gdna
‑
ha模板(100ng至100pg)、1
×
ddpcr
tm
探针超混合物(无dutp)(bio
‑
rad公司,加州,美国)。制备ddpcr液滴混合物,并如上所述进行ddpcr反应。
[0099]
1.5构筑带有ha
‑
n myc
‑
ha以及ha
‑
nagk
‑
ha序列的载体以作为分析标准
[0100]
然后使用he swift选殖试剂盒(toolbiotech公司,中国台湾)选殖ha
‑
n myc
‑
ha以及ha
‑
nagk
‑
ha构筑体,然后转化为dh5α胜任细胞,并铺在胺芐青霉素lb琼脂培养盘上。筛选细菌菌落并通过sanger定序法定序,以使用qiaprep spin miniprep试剂盒(qiagen公司,nw,德国)在质体萃取后验证序列。
[0101]
ddtop
‑
pcr的标准模板是通过在100μl pcr反应中扩增具有正确ha
‑
nmyc
‑
ha以及ha
‑
nagk
‑
ha序列的重组质体而产生的,该pcr反应含有phe
‑
f引子(5
’‑
cga ctc act ata ggg aga gcg gc
‑3’
;seq id no:17,0.5μm)、phe
‑
r引子(5
’‑
aa gaa cat cga ttt tcc atg gca g
‑3’
;seq id no:18,0.5μm)、dna(1ng)、1x q5 high
‑
fidelity master mix。然后将混合物于98℃下作用1分钟,然后于98℃作用20秒,64℃作用30秒,以及72℃作用10秒,共30个循环。混合物随后在72℃下作用2分钟以进行最终延伸。使用qiaquick pcr纯化试剂盒(qiagen公司,nw,德国)纯化大小分别为309bp以及325bp的phe
‑
ha
‑
n myc
‑
ha与phe
‑
ha
‑
nagk
‑
ha pcr扩增子并定量。
[0102]
1.6构筑5'缺失的n
‑
myc以及nagk构筑体以作为分析标准
[0103]
n
‑
myc的5'引子结合位点缺失的构筑体以正向引子(5
’‑
agc gct aga ctc cga ctt cac taa agt tcc ttc cac cct ctc ctg ggg ag
‑3’
)(seq id no:19)以及反向引子(5
’‑
agc gct aga ctc cga ctt agc cac caa ctt tct cca att tta ttc ctc ag
‑3’
)(seq id no:20)由q5 high
‑
fidelity master mix进行扩增。nagk的5'引子结合位点缺失的构筑体以正向引子(5
’‑
agc gct aga ctc cga ctg tgt tgc ccg aga ttg acc cgg tga gtt gag gt
‑3’
)(seq id no:21)以及反向引子(5
’‑
agc gct aga ctc cga cta tgc agg gtt tga tgg gat agt ccc atc
‑3’
)(seq id no:22)由q5 high
‑
fidelity master mix进行扩增。大小分别为160bp以及146bp的ha
‑
n myc
‑
f del
‑
ha以及ha
‑
nagk
‑
f del
‑
ha pcr扩增子以qiaquick pcr纯化试剂盒(qiagen公司,nw,德国)纯化并定量。
[0104]
1.7用于扩增的pcr引子
[0105]
从lodrini等人得到ddpcr的配对引子序列,但其序列略有修饰(lodrini等人,2017年)。引子与探针由integrated dna technology公司(idt)合成。实际上,为了扩增n
‑
myc序列,使用正向引子(5
’‑
gtg ctc tcc aat tct cgc ct
‑3’
)(seq id no:3)以及反向引子(5
’‑
gat ggc cta gag gag ggc t
‑3’
)(seq id no:4)。
[0106]
1.8检测用探针
[0107]
为了检测n
‑
myc扩增,设计了3个探针(如下所示)。这些包括1)探针mpb1(fam
‑
n
‑
myc探针):/56
‑
fam/cac taa agt/zen/tcc ttc cac cct ctc ct/3iabkfq/(seq id no:10);2)探针mpb1 1(fam
‑
n
‑
myc探针 1nt):/56
‑
fam/cac taa agt/zen/tcc ttc cac cct ctc ctg/3iabkfq/(seq id no:11);3)探针mpb1 2(fam
‑
n
‑
myc探针 2nt):/56
‑
fam/cac taa agt/zen/tcc ttc cac cct ctc ctg g/3iabkfq/(seq id no:12)。该初始测试使用的是探针mpb1 1。
[0108]
1.9ddpcr与ddtop
‑
pcr的条件
[0109]
为了实现ddtop
‑
pcr的可行状态并最终优化其灵敏度及特异性,必须测试pcr引子及荧光探针的长度以及实验条件。此外,必需同时运行传统ddpcr以作为初始设置的阳性对照,因此优化及实验条件都应用于ddtop
‑
pcr与ddpcr。
[0110]
根据初步结果,我们确定对基因特异性引子(ddpcr对照)使用浓度为0.9μm的引子,对t/u寡核苷酸(ddtop
‑
pcr)使用浓度为32μm的引子。总共20μl pcr反应包含一或多个引子(用于ddpcr对照的配对引子或用于ddtop
‑
pcr的t/u寡核苷酸引子)、0.25μm探针、dna模板(2.0μl),1
×
ddpcr探针超混合物(无dutp)(bio
‑
rad公司)。通过将制备的pcr反应混合物(20μl)与70μl液滴数位pcr油(bio
‑
rad公司)混合来产生液滴。将总共40μl ddpcr液滴混合物转移至96孔盘,并在以bio
‑
rad t
‑
100热循环仪(bio
‑
rad公司)进行pcr反应之前密封。然后将ddpcr以及ddtop
‑
pcr制备物都置于bio
‑
rad pcr仪中,用于在以下条件下扩增并产生发色团:95℃作用10分钟,然后于94℃作用30秒以及58℃作用60秒共40个循环。通过在98℃下作用10分钟来终止反应。pcr反应后,使用qx200 ddpcr液滴读取仪进行阳性液滴的定量,并使用quantasoft分析软件(版本1.7.4,bio
‑
rad公司)进行分析。
[0111]
2.结果
[0112]
2.1使用ddpcr以及ddtop
‑
pcr检测n
‑
myc基因序列进行比较
[0113]
进行最初的测试以证明此一概念,即通过以top
‑
pcr代替传统pcr,应当能够提高灵敏度,这表示为qx200 ddpcr仪中阳性计数的增加。
[0114]
仅包含nagk或n
‑
myc序列的样本提供了易于验证的系统来改善条件,在此条件下可以根据实验结果调整实验条件(例如,样本制备、pcr成分浓度,以及反应条件)。
[0115]
为了进行测试,我们准备了两种类型的含n
‑
myc序列的片段:一种带有两个引子结合位点,而另一种则缺失了5'引子结合位点,仅保留了3'结合位点,还有nagk缺失5'引子结合位点的对照(表1)。
[0116]
表1.实验设计
[0117][0118][0119]
我们将qx200 ddpcr仪用于不同的程序,ddpcr或ddtop
‑
pcr,以证明ddtop
‑
pcr在检测有缺陷引子结合位点的片段中是具有潜力的,这些片段可能存在于cfna样本池中。
[0120]
对于概念验证测试,我们准备了包含可变百分比的上述5'引子结合位点缺失模板的样本。计算得出每个样本的初始输入总量约为12,000个复制,所有实验均由qx200 ddpcr仪使用lodrini等人报告的设置进行,但进行了小部分修改(lodrini等人,2017年)。lodrini等人使用的相同的n
‑
myc寡核苷酸探针序列也用于该实验。
[0121]
结果显示,一般而言,使用t寡核苷酸作为用于扩增的唯一引子的ddtop
‑
pcr方法比使用双重内部引子的ddpcr的计数更高(图3)。
[0122]
为了比较ddtop
‑
pcr与ddpcr的灵敏度,我们估算了原始样本中的复制数输入(使用qubit)以及相应检测出的复制数,然后计算出检测出的百分比(表2)。
[0123]
表2.ddpcr以及ddtop
‑
pcr之间的灵敏度比较。
[0124][0125]
结果显示,ddpcr能够检测到约48.6%的模板,而ddtop
‑
pcr能够检测到约58.5%
‑
62.8%的模板,这表示从ddpcr到ddtop
‑
pcr的灵敏度提高了10%
‑
15%。请注意,尽管准确性受量化设备(例如qubit)、个人技术、qx200机器本身等因素造成的偏差/变化的影响,但每种方法的总体趋势都具有一定程度的可靠性。
[0126]
计算标准偏差
[0127]
但是,ddtop
‑
pcr的更高灵敏度受到更多分散信号强度的影响(图4)。ddtop
‑
pcr液滴中的大多数阳性信号强度均低于ddpcr液滴。推测是由于使用双重内部引子导致较短而明确的扩增范围,进而使ddpcr产生比ddtop
‑
pcr更高均匀度的信号,而ddtop
‑
pcr的扩增始于侧翼衔接子,其远离双引子位置。
[0128]
如图4的所有混合样本所示,从ddtop
‑
pcr产生的液滴的颜色强度可以比从ddpcr产生的液滴的颜色强度更高或更低,并且更分散。如第4道所示,ddpcr无法检测到5'引子结合位点缺失的片段,而ddtop
‑
pcr却能高效检测到该片段。图中的前两个泳道表示nagk模板的伪阳性较低。先前的观察结果显示,对于ddtop
‑
pcr以及ddpcr方法,空白背景均具有干净计数(0)(数据未显示)。
[0129]
如ddpcr所示,内部双引子的扩增产生更均一的结果,而另一方面,基于衔接子的ddtop
‑
pcr的扩增具有更高的灵敏度,但强度较低。
[0130]
这些数据还表示需要进一步优化。而且,较短的片段似乎比较长的片段具有优势。
[0131]
3.结论
[0132]
这项概念验证研究提供了初步数据来证明ddtop
‑
pcr的发展,并通过实验证明使用ddtop
‑
pcr来提高基于cfdna的复制数变异(cnv)、基因突变以及疾病基因表现以及snp改变的分析的准确性的可行性。
[0133]
相较于传统的ddpcr,ddtop
‑
pcr具有许多优点:1)数位pcr不适合cfdna分析,因为作为模板依赖性方法,传统pcr需要两个引子结合位点共同存在于同一片段中。另一方面,作为依赖于衔接子的pcr方法,ddtop
‑
pcr没有这种限制,因此能够检测部分片段。2)在
citri subsp.citri.plos one 11,e0159004.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。