1.本发明属于有机化学合成技术领域,尤其涉及一种光学纯的手性氨基缩醛及其制备方法与应用。
背景技术:
2.手性α
‑
氨基缩醛作为一类新的手性α
‑
氨基酸衍生物,广泛的存在于各类天然产物和手性药物当中,并且由于其相邻的两个碳原子分别具有缩醛和氨基官能团的独特结构使其成为重要的药物及天然产物合成中间体。手性α
‑
氨基酸衍生物的种类很多,其中最具代表性的有α
‑
氨基醛、α
‑
氨基醇、α
‑
氨基酰胺、α
‑
氨基羧酸酯等,这些类手性氨基酸衍生物均可以通过手性α
‑
氨基缩醛简单转化和修饰获得。基于这类α
‑
氨基缩醛在生命科学研究、有机合成以及生物医药研发当中的潜在价值,在过去几十年中已经引起了众多合成化学家们的浓厚兴趣。在医药领域和众多的天然产物当中,有一半以上的药物和天然产物是手性分子。因此,合成一系列的手性氨基酸衍生物并应用于构建手性药物和天然产物骨架是当今合成化学中热门的课题之一。
3.目前,酶促动力学拆分、通过以非对映异构体成盐的方式进行的经典拆分以及不对称相转移催化反应是最常用的合成方法,并实现了工业化生产。但是此类合成方法存在酶易失活,产物易消旋化,分离效果不好,原子利用率低,反应条件苛刻,获得的对映体选择性尚待提高等缺陷。此外,对天然α
‑
氨基酸衍生物经c
‑
h官能化进行修饰也是当今合成化学家们研究的热门领域,然而,天然氨基酸数量有限而且底物需要进行多次上保护基及脱保护基操作。
4.因此,利用商业化的脂肪醛、二级胺为原料制备多样化的α
‑
氨基缩醛,进而动力学拆分制备光学纯的α
‑
氨基缩醛具有重要的意义及应用价值。
技术实现要素:
5.本发明的首要目的在于提供一种光学纯的手性氨基缩醛的制备方法,旨在克服上述背景技术中现有技术的不足之处。
6.本发明的再一目的在于提供了由上述制备方法得到的光学纯的手性氨基缩醛。
7.本发明的另一目的在于提供上述光学纯的手性氨基缩醛的应用。
8.本发明是这样实现的,一种光学纯的手性氨基缩醛的制备方法,该方法包括以下步骤:
9.(1)将0.2~3mmol外消旋的对硝基苯磺酰氨基缩醛、0.2~3mmol芳香基频那醇酯、0.02~0.3mmol醋酸钯、0.03~0.45mmol手性氨基酸配体、0.6~9mmol碳酸钠、0.4~6mmol碳酸银、0.1~1.5mmol 1,4
‑
苯醌、0.08~1.2mmol二甲基亚砜、1~15mmol水以及1~15ml叔戊醇加入到反应容器中,混合均匀得到混合物,将该混合物在惰性气体环境和50℃温度下反应6~12小时,得到反应产物;
10.(2)将所述反应产物进行纯化,得到光学纯的手性氨基缩醛。
11.优选地,在步骤(1)中,所述外消旋的对硝基苯磺酰氨基缩醛选自n
‑
(2,2
‑
二甲氧基
‑1‑
苯基乙基)
‑4‑
硝基苯磺酰胺、n
‑
(2,2
‑
二甲氧基
‑1‑
(邻甲苯基)乙基)
‑4‑
硝基苯磺酰胺、n
‑
(2,2
‑
二甲氧基
‑1‑
(2
‑
甲氧基苯基)乙基)
‑4‑
硝基苯磺酰胺、n
‑
(1
‑
(2
‑
氟苯基)
‑
2,2
‑
二甲氧基乙基)
‑4‑
硝基苯磺酰胺、n
‑
(2,2
‑
二甲氧基
‑1‑
(2
‑
(三氟甲基)苯基)乙基)
‑4‑
硝基苯磺酰胺、n
‑
(1
‑
(3
‑
氟苯基)
‑
2,2
‑
二甲氧基乙基)
‑4‑
硝基苯磺酰胺、n
‑
(2,2
‑
二甲氧基
‑1‑
(3
‑
甲氧基苯基)乙基)
‑4‑
硝基苯磺酰胺、n
‑
(2,2
‑
二甲氧基
‑1‑
(3
‑
(三氟甲基)苯基)乙基)
‑4‑
硝基苯磺酰胺、n
‑
(1
‑
(2
‑
,3
‑
二氟苯基)
‑
2,2
‑
二甲氧基乙基)
‑4‑
硝基苯磺酰胺、n
‑
(1
‑
(2,3,4
‑
三氟苯基)
‑
2,2
‑
二甲氧基乙基)
‑4‑
硝基苯磺酰胺、n
‑
(3
‑
(2
‑
氟苯基)
‑
1,1
‑
二甲氧基丙烷
‑2‑
基)
‑4‑
硝基苯磺酰胺以及n
‑
(1
‑
(苯并[d][1,3]二氧杂
‑5‑
基)
‑
2,2
‑
二甲氧基乙基)
‑4‑
硝基苯磺酰胺中的任意一种。
[0012]
优选地,在步骤(1)中,所述外消旋的对硝基苯磺酰氨基缩醛的制备过程为:
[0013]
a、将0.5~5mmol二苄胺、0.6~6mmol脂肪醛、0.75~7.5mmol过碳酸钠、0.1~1mmol碘、0.5~5ml甲醇以及2~20ml1,2
‑
二氯乙烷加入到反应容器中,混合均匀得到混合物1,将该混合物1在40~60℃温度下反应6~12小时,得到二苄基氨基缩醛;
[0014]
b、将步骤a中得到的0.5~5mmol二苄氨基缩醛、pd(oh)2/c(10wt%),以及0.5~5ml甲醇加入到反应容器中,在20~50℃下搅拌12~24小时,过滤得到脱苄基的氨基缩醛;
[0015]
c、将步骤b中得到的0.5~5mmol脱苄基的氨基缩醛、0.5~5mmol对硝基苯磺酰氯、1.5~15mmol三乙胺以及2~20ml二氯甲烷加入到反应容器中,混合均匀得到混合物2,将该混合物2在0~25℃温度下反应5~12小时,得到外消旋的对硝基苯磺酰氨基缩醛。
[0016]
优选地,在步骤(1)中,所述芳香基频那醇酯选自苯硼酸频那醇酯、4
‑
酯基苯硼酸频那醇酯、4
‑
氟苯硼酸频那醇酯、4
‑
氯基苯硼酸频那醇酯、4
‑
三氟甲基苯硼酸频那醇酯、4
‑
氰基苯硼酸频那醇酯、3
‑
酯基苯硼酸频那醇酯、2
‑
氟
‑3‑
氰基苯硼酸频那醇酯中的任意一种。
[0017]
优选地,在步骤(1)中,所述惰性气体为氮气。
[0018]
优选地,在步骤(2)中,将所述反应产物通过薄层层析法进行纯化,展开剂体系为石油醚/乙酸乙酯,且石油醚和乙酸乙酯用量比为10~5:1。
[0019]
本发明进一步公开了由上述制备方法得到的手性氨基缩醛,该手性氨基缩醛的化学式如下式(ⅰ)、式(ⅱ)或式(ⅲ)所示:
[0020][0021][0022]
式(i)、式(ⅱ)以及式(ⅲ)中,r1选自2
‑
me、2
‑
ome、2
‑
f、2
‑
cf3、3
‑
f、3
‑
ome、3
‑
cf3、2,3
‑
2f、4
‑
f、4
‑
cf3、4
‑
ome、
‑
och2o
‑
中的任意一种;r3选自me、et、ac中的任意一种;
[0023]
式(ⅱ)以及式(ⅲ)中,r2选自h、4
‑
co2me、4
‑
f、4
‑
cl、4
‑
cf3、4
‑
cn、3
‑
co2me、3
‑
cn、2
‑
f
‑3‑
cn中的任意一种;
[0024]
或者,该手性氨基缩醛的化学式如下式(ⅳ)、式(
ⅴ
)或式(
ⅵ
)所示:
[0025][0026]
式(ⅳ)、式(
ⅴ
)以及式(
ⅵ
)中,r4选自2
‑
f、2
‑
cf3、3
‑
ome、2,3
‑
2f、中的任意一种;r5选自me、et、ac中的任意一种;
[0027]
式(
ⅴ
)以及式(
ⅵ
)中,r6选自h、4
‑
co2me、4
‑
f、4
‑
cl、4
‑
cf3、4
‑
cn、3
‑
co2me、3
‑
cn、2
‑
f
‑3‑
cn中的任意一种。
[0028]
本发明进一步公开了上述手性氨基缩醛做为中间体在制备药物以及合成天然产物中的应用。
[0029]
相比于现有技术的缺点和不足,本发明具有以下有益效果:
[0030]
(1)本发明方法所用的氨基缩醛为合成简单、转化率高的脂肪醛氨基化反应制备,底物适用范围广,适用于多种取代基的芳香基类氨基缩醛,并且催化体系中使用的钯催化剂以及手性氨基酸配体比较廉价,经济性高、市售来源广的特点;并且本发明方法所需反应条件非常温和,反应步骤少,具有操作简单的特点;
[0031]
(2)本发明制备的光学纯的α
‑
氨基缩醛作为一类重要的手性α
‑
氨基酸的衍生物,广泛分布在生物学和药学活性分子中,经简单转化能实现多类手性氮杂环的合成,在药物及天然产物合成中具有广阔的应用前景。
附图说明
[0032]
图1是本发明实施例中外消旋的n
‑
(2,2
‑
二甲氧基
‑1‑
(邻甲苯基)乙基)
‑4‑
硝基苯磺酰胺的核磁共振氢谱图;
[0033]
图2是本发明实施例中外消旋的n
‑
(2,2
‑
二甲氧基
‑1‑
(邻甲苯基)乙基)
‑4‑
硝基苯磺酰胺的核磁共振碳谱图;
[0034]
图3是本发明实施例中外消旋的n
‑
(2,2
‑
二甲氧基
‑1‑
(邻甲氧基苯基)乙基)
‑4‑
硝基苯磺酰胺的核磁共振氢谱图;
[0035]
图4是本发明实施例中外消旋的n
‑
(2,2
‑
二甲氧基
‑1‑
(邻甲氧基苯基)乙基)
‑4‑
硝基苯磺酰胺的核磁共振碳谱图;
[0036]
图5是本发明实施例中外消旋的n
‑
(3
‑
(2
‑
氟苯基)
‑
1,1
‑
二甲氧基丙烷
‑2‑
基)
‑4‑
硝基苯磺酰胺的核磁共振氢谱图;
[0037]
图6是本发明实施例中外消旋的n
‑
(3
‑
(2
‑
氟苯基)
‑
1,1
‑
二甲氧基丙烷
‑2‑
基)
‑4‑
硝基苯磺酰胺的核磁共振碳谱图;
[0038]
图7是本发明实施例中外消旋的n
‑
(3
‑
(2
‑
氟苯基)
‑
1,1
‑
二甲氧基丙烷
‑2‑
基)
‑4‑
硝基苯磺酰胺的核磁共振氟谱图;
[0039]
图8是本发明实施例中(s)
‑
2'
‑
(2,2
‑
二甲氧基
‑1‑
(((4
‑
硝基苯基)磺酰胺基)乙基)
‑
3'
‑
甲基
‑
[1,1'
‑
联苯]
‑4‑
羧酸甲酯的核磁共振氢谱图;
[0040]
图9是本发明实施例中2'
‑
(2,2
‑
二甲氧基
‑1‑
(((4
‑
硝基苯基)磺酰胺基)乙基)
‑
3'
‑
甲基
‑
[1,1'
‑
联苯]
‑4‑
羧酸甲酯的核磁共振碳谱图;
[0041]
图10是本发明实施例中(s)
‑
2'
‑
(2,2
‑
二甲氧基
‑1‑
(((4
‑
硝基苯基)磺酰胺基)乙基)
‑
3'
‑
甲氧基
‑
[1,1'
‑
联苯]
‑4‑
羧酸甲酯的核磁共振氢谱图;
[0042]
图11是本发明实施例中2'
‑
(2,2
‑
二甲氧基
‑1‑
(((4
‑
硝基苯基)磺酰胺基)乙基)
‑
3'
‑
甲氧基
‑
[1,1'
‑
联苯]
‑4‑
羧酸甲酯的核磁共振碳谱图;
[0043]
图12是本发明实施例中(s)
‑
n
‑
(3
‑
(3
‑
氟
‑
[1,1'
‑
联苯]
‑2‑
基)
‑
1,1
‑
二甲氧基丙烷
‑2‑
基)
‑4‑
硝基苯磺酰胺的核磁共振氢谱图;
[0044]
图13是本发明实施例中(s)
‑
n
‑
(3
‑
(3
‑
氟
‑
[1,1'
‑
联苯]
‑2‑
基)
‑
1,1
‑
二甲氧基丙烷
‑2‑
基)
‑4‑
硝基苯磺酰胺的核磁共振碳谱图;
[0045]
图14是本发明实施例中(s)
‑
n
‑
(3
‑
(3
‑
氟
‑
[1,1'
‑
联苯]
‑2‑
基)
‑
1,1
‑
二甲氧基丙烷
‑2‑
基)
‑4‑
硝基苯磺酰胺的核磁共振氟谱图。
具体实施方式
[0046]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0047]
实施例1
[0048]
a、将1.0mmol二苄胺、1.2mmol邻甲基苯乙醛、1.5mmol过碳酸钠、0.2mmol碘、1.0ml甲醇以及4ml 1,2
‑
二氯乙烷加入到反应容器中,混合均匀得到混合物1,将该混合物1在60℃温度下反应12小时,得到二苄基氨基缩醛;
[0049]
b、将步骤a中得到的0.5mmol二苄氨基缩醛、pd(oh)2/c(10wt%),以及0.5ml甲醇加入到反应容器中,在50℃下搅拌12小时,过滤得到脱苄基的氨基缩醛;
[0050]
c、将步骤b中得到的0.5mmol脱苄基的氨基缩醛、0.5mmol对硝基苯磺酰氯、1.5mmol三乙胺以及2ml二氯甲烷加入到反应容器中,混合均匀得到混合物2,将该混合物2在0~25℃温度下反应12小时,得到外消旋的n
‑
(2,2
‑
二甲氧基
‑1‑
(邻甲苯基)乙基)
‑4‑
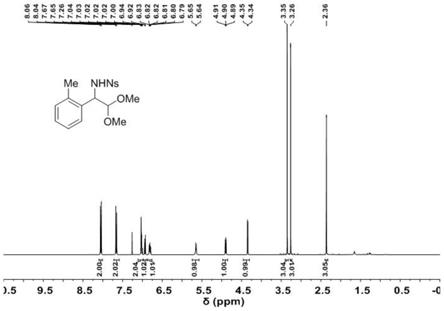
硝基苯磺酰胺。该浅黄色固体的化学结构及核磁共振图如图1~2所示。其核磁数据表征归属如下:r
f
=0.35(hexane:ethyl acetate=3:1);1h nmr(400mhz,cdcl3)δ8.05(d,j=8.9hz,2h),7.66(d,j=8.9hz,2h),7.02(m,2h),6.93(d,j=7.6hz,1h),6.81(m,1h),5.65(d,j=5.8hz,1h),4.90(m,1h),4.35(j=4.7hz,1h),3.35(s,3h),3.26(s,3h),2.36(s,3h)ppm;
13
c nmr(100mhz,cdcl3)ppm;δ149.41,146.40,136.35,134.06,130.21,128.09,127.78,127.49,125.81,123.48,105.78,56.09,55.21,55.07,19.40;hrms(esi
‑
tof)m/z calcd for c
16
h
17
n2o6sh
[m h]
381.1120,found 381.1126.
[0051]
实施例2
[0052]
a、将1.0mmol二苄胺、1.2mmol邻甲氧基苯乙醛、1.5mmol过碳酸钠、0.2mmol碘、1.0ml甲醇以及4ml 1,2
‑
二氯乙烷加入到反应容器中,混合均匀得到混合物1,将该混合物1在60℃温度下反应12小时,得到二苄基氨基缩醛;
[0053]
b、将步骤a中得到的0.5mmol二苄氨基缩醛、pd(oh)2/c(10wt%),以及0.5ml甲醇加入到反应容器中,在50℃下搅拌12小时,过滤得到脱苄基的氨基缩醛;
[0054]
c、将步骤b中得到的0.5mmol脱苄基的氨基缩醛、0.5mmol对硝基苯磺酰氯、1.5mmol三乙胺以及2ml二氯甲烷加入到反应容器中,混合均匀得到混合物2,将该混合物2在0~25℃温度下反应12小时,得到外消旋的n
‑
(2,2
‑
二甲氧基
‑1‑
(邻甲氧基苯基)乙基)
‑4‑
硝基苯磺酰胺。该浅黄色固体的化学结构及核磁共振图如图3~4所示。其核磁数据表征归属如下:r
f
=0.35(he xane:ethyl acetate=3:1);1h nmr(400mhz,cdcl3)δ8.10(d,j=7.8hz,2h),7.82(d,j=8.6hz,2h),7.14(t,j=7.8hz,1h),7.07(d,j=7.5hz,1h),6.76(t,j=7.5hz,1h),6.68(d,j=8.3hz,1h),5.83(m,1h),4.82(m,1h),4.46(d,j=4.7hz,1h),3.76(s,3h),3.28(s,3h),3.20(s,3h)ppm;
13
c nmr(100mhz,cdcl3)δ156.29,149.56,146.63,129.63,129.33,128.36,124.42,123.56,120.73,110.43,104.35,55.92,55.45,55.16,55.07ppm;hrms(esi
‑
tof)m/z calcd for c
17
h
20
n2o7sh
[m h]
397.1069,found 397.1063.
[0055]
实施例3
[0056]
a、将1.0mmol二苄胺、1.2mmol邻氟苯丙醛、1.5mmol过碳酸钠、0.2mmol碘、1.0ml甲醇以及4ml 1,2
‑
二氯乙烷加入到反应容器中,混合均匀得到混合物1,将该混合物1在60℃温度下反应12小时,得到二苄基氨基缩醛;
[0057]
b、将步骤a中得到的0.5mmol二苄氨基缩醛、pd(oh)2/c(10wt%),以及0.5ml甲醇加入到反应容器中,在50℃下搅拌12小时,过滤得到脱苄基的氨基缩醛;
[0058]
c、将步骤b中得到的0.5mmol脱苄基的氨基缩醛、0.5mmol对硝基苯磺酰氯、1.5mmol三乙胺以及2ml二氯甲烷加入到反应容器中,混合均匀得到混合物2,将该混合物2在0~25℃温度下反应12小时,得到外消旋的n
‑
(3
‑
(2
‑
氟苯基)
‑
1,1
‑
二甲氧基丙烷
‑2‑
基)
‑4‑
硝基苯磺酰胺。该浅黄色固体的化学结构及核磁共振图如图5~7所示。其核磁数据表征归属如下:r
f
=0.35(hexane:ethyl acetate=3:1);1h nmr(400mhz,cdcl3)δ8.07(d,j=8.6hz,2h),7.69(d,j=8.8hz,2h),7.07(m,1h),6.98(m,1h),6.87(td,j=7.5hz,1hz,1h),6.75(m,1h),5.03(d,j=9.2hz,1h),4.34(d,j=2.6hz,1h),3.65(m,1h),3.49(s,3h),3.42(s,3h),2.93(dd,j=14.2hz,4.6hz,1h),2.70(dd,j=14.3hz,10.2hz,1h);
13
c nmr(100mh z,cdcl3):δ162.27,159.84,149.44,146.10,131.69(d,j=4.8hz),129.21128.50(d,j=1.7hz),128.41,127.84,127.43,124.50,124.35,124.12(d,j=3.6hz),123.83,115.16(d,j=22.1hz),106.17,99.87,57.27,56.75,28.49(d,j=1.8hz)ppm;hrms(esi
‑
tof)m/z calcd for c
17
h
19
fn2o6s h
[m h]
399.1026,found 399.1030.
[0059]
实施例4~10
[0060]
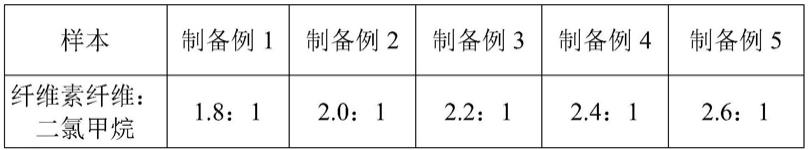
实施例4~10与实施例1相似,差别之处在于步骤a中脂肪醛的选择不同,具体如下表1所示:
[0061]
表1
[0062]
[0063][0064]
实施例11
[0065]
(1)在10ml史莱克管中,在氮气环境下,依次加入外消旋的n
‑
(2,2
‑
二甲氧基
‑1‑
(邻甲苯基)乙基)
‑4‑
硝基苯磺酰胺(0.10mmol,0.046g)、4
‑
酯基苯硼酸频那醇酯(0.1mmol,0.035g)、醋酸钯(0.01mmol,0.0023g)、手性氨基酸配体(0.015mmol,0.0056g)以及碳酸钠(0.3mmol,0.033g)、碳酸银(0.2mmol,0.0553g)、1,4
‑
苯醌(0.05mmol,0.056g)、二甲基亚砜(0.04mmol,0.003ml)、水(0.5mmol,0.01ml)50℃下在0.5ml叔戊醇中搅拌反应6小时,反应方程式为:
[0066][0067]
(2)tlc监测反应完成后,用二氯甲烷将混合物溶解,薄层层析法(石油醚/乙酸乙酯=5:1)分离产物,产物为浅黄色固体状化合物1,收率51%。该浅黄色固体的化学结构及核磁共振图如图8~9所示。
[0068]
浅黄色固体;r
f
=0.33(hexane:ethyl acetate=8:1);1h nmr(400m hz,cdcl3)δ8.12(m,4h),7.64(d,j=8.6hz,2h),7.41(s,1h),7.19(t,j=7.6hz,1h),7.01(dd,j=21.9hz,7.2hz,2h),5.40(s,1h),4.75(s,1h),4.45(s,1h),3.97(s,3h),3.14(s,3h),2.92(s,3h),2.41(s,3h);
13
c nmr(100mhz,cdcl3):δ166.78,149.52,146.35,137.32,137.22,
128.99,128.22,127.72,123.59,104.46,55.78,55.67,53.56,52.27,21.23.hrms(esi
‑
tof)m/z calcd for c
25
h
26
n2o8sh
[m h]
515.1488,found 515.1490.hplc,chiralcel ad
‑
h column(20%isopropanol in hexanes,0.8ml/min)tr 13.12min(major),22.46min(minor):92%ee.
[0069]
实施例12
[0070]
(1)在10ml史莱克管中,在氮气环境下,依次加入外消旋的n
‑
(2,2
‑
二甲氧基
‑1‑
(邻甲氧基苯基)乙基)
‑4‑
硝基苯磺酰胺(0.10mmol,0.040g)、4
‑
酯基苯硼酸频那醇酯(0.1mmol,0.035g)、醋酸钯(0.01mmol,0.0023g)、手性氨基酸配体(0.015mmol,0.0056g)以及碳酸钠(0.3mmol,0.033g)、碳酸银(0.2mmol,0.0553g)、1,4
‑
苯醌(0.05mmol,0.056g)、二甲基亚砜(0.04mmol,0.003ml)、水(0.5mmol,0.01ml)50℃下在0.5ml叔戊醇中搅拌反应6小时,反应方程式为:
[0071][0072]
(2)tlc监测反应完全后,用二氯甲烷将混合物取出,薄层层析法(石油醚:乙酸乙酯=5:1)分离产物,产物为淡黄色液体状化合物2,收率47%。该淡黄色固体的化学结构及核磁共振图如图10~11所示。
[0073]
浅黄色固体;mp 137.9
‑
138.8℃;r
f
=0.33(hexane:ethyl acetate=8:1);1h nmr(400mhz,cdcl3)δ8.10(dd,j=12.7hz,8.5hz,4h),7.67(d,j=8.9hz,2h),7.44(s,1h),7.23(d,j=7.9hz,1h),7.79(dd,j=8.1hz,1.6hz,2h),6.13(d,j=9.2hz,1h),4.69(m,2h),3.96(s,3h),3.84(s,3h),3.10(s,3h),3.03(s,3h);
13
c nmr(100mhz,cdcl3):δ166.96,156.99,149.37,146.77,144.72,143.06,129.18,129.07,128.91,128.04,123.45,110.30,104.22,56.21,55.64,55.00,52.52,52.21.hrms(esi
‑
tof)m/z calcd for c
25
h
26
n2o9sh
[m h]
531.1437,found 531.1435.
[0074]
hplc,chiralcel ad
‑
h column(20%isopropanol in hexanes,0.8ml/min)tr 18.5min(major),31.4min(minor):94%ee.
[0075]
实施例13
[0076]
(1)在10ml史莱克管中,在氮气环境下,依次加入外消旋的n
‑
(3
‑
(2
‑
氟苯基)
‑
1,1
‑
二甲氧基丙烷
‑2‑
基)
‑4‑
硝基苯磺酰胺(0.10mmol,0.040g)、苯硼酸频那醇酯(0.1mmol,0.024g)、醋酸钯(0.01mmol,0.0023g)、手性氨基酸配体(0.015mmol,0.0056g)以及碳酸钠(0.3mmol,0.033g)、碳酸银(0.2mmol,0.0553g)、1,4
‑
苯醌(0.05mmol,0.056g)、二甲基亚砜(0.04mmol,0.003ml)、水(0.5mmol,0.01ml)50℃下在1.5ml叔戊醇中搅拌反应6小时,反应方程式为:
[0077][0078]
(2)tlc监测反应完全后,用二氯甲烷将混合物取出,薄层层析法(石油醚:乙酸乙酯=5:1)分离产物,产物为淡黄色固体状化合物3,收率48%。该淡黄色固体的化学结构及核磁共振图如图12~14所示。
[0079]
浅黄色固体;r
f
=0.33(hexane:ethyl acetate=8:1);1h nmr(400mhz,cdcl3)δ8.12(d,j=9.0hz,2h),7.67(d,j=8.9hz,2h),7.42(m,3h),7.13(m,2h),7.08(m,1h),6.84(d,j=7.6hz,1h),6.76(m,1h),4.80(d,j=9.5hz,1h),4.05(d,j=2.6hz,1h),3.36(m,1h),3.23(s,3h),3.08(s,3h),2.93(m,1h),2.82(m,1h);
13
c nmr(100mhz,cdcl3):δ162.57,160.14,149.42,146.44,144.55(d,j=4.2hz),139.66(d,j=2.9hz),128.92,128.52,127.75,127.66,127.62,127.59,125.83(d,j=3.2hz),123.84,122.77,122.62,114.23,114.00,106.03,56.33,56.02,55.74(d,j=1.9hz),23.98(d,j=2.9hz).
[0080]
hplc,chiralcel ad
‑
h column(20%isopropanol in hexanes,0.8ml/min)tr 12.5min(major),17.0min(minor):91%ee.
[0081]
实施例14
[0082]
(1)在10ml史莱克管中,在氮气环境下,依次加入(r)
‑
n
‑
苄基
‑
n
‑
(2,2
‑
二甲氧基
‑1‑
(2
‑
(三氟甲基)苯基)乙基)
‑4‑
硝基苯磺酰胺(化合物a)、fecl3(0.2mmol,0.033g),二氯甲烷作溶剂,在
‑
20℃反应条件下搅拌反应0.5小时,反应方程式为:
[0083][0084]
(2)tlc检测反应完成后,减压蒸馏除去溶剂,薄层层析法(石油醚:乙酸乙酯=10:1)分离产物,产物为浅黄色固体状化合物b,收率88%。
[0085]
实施例15
[0086]
(1)在10ml试管中,依次加入(r)
‑
n
‑
(2,2
‑
二甲氧基
‑1‑
苯乙基)
‑4‑
硝基
‑
n
‑
(2
‑
苯基烯丙基)苯磺酰胺(化合物c)、tfoh(0.2mmol,0.031g),二氯甲烷作溶剂,在20℃反应条件下搅拌反应0.5小时,反应方程式为:
[0087][0088]
(2)tlc检测反应完成后,减压蒸馏除去溶剂,薄层层析法(石油醚:乙酸乙酯=10:
1)分离产物,产物为白色固体状化合物d,收率90%。
[0089]
实施例16
[0090]
(1)在10ml史莱克管中,在氮气环境下,依次加入(r)
‑
n
‑
(2,2
‑
二甲氧基
‑1‑
(2
‑
(三氟甲基)苯基)乙基)
‑
n
‑
(3
‑
甲基
‑2‑
烯
‑1‑
基)
‑4‑
硝基苯磺酰胺(化合物e)、tfoh(0.2mmol,0.031g),二氯甲烷作溶剂,在0℃反应条件下搅拌反应0.5小时反应方程式为:
[0091][0092]
(2)tlc监测反应完成后,用二氯甲烷将混合物溶解,薄层层析法(石油醚/乙酸乙酯=10:1)分离产物,产物为白色固体状化合物f,收率89%。
[0093]
实施例17
[0094]
(1)在10ml史莱克管中,在氮气环境下,依次加入(r)
‑
n
‑
(2,2
‑
二甲氧基
‑1‑
(3
‑
(三氟甲基)
‑
[1,1'
‑
联苯]
‑2‑
基)乙基)
‑4‑
硝基苯磺酰胺(化合物g)、fecl3(0.2mmol,0.033g),二氯甲烷作溶剂,在
‑
20℃反应条件下搅拌反应0.5小时反应方程式为:
[0095][0096]
(2)tlc监测反应完成后,用二氯甲烷将混合物溶解,薄层层析法(石油醚/乙酸乙酯=10:1)分离产物,产物为浅黄色固体状化合物h,收率80%。
[0097]
实施例18~25
[0098]
本实施例18~25与上述实施例11基本相同,差别之处如下表2所示:
[0099]
表2差别比较
[0100][0101]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。