一种2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶的合成方法
技术领域
1.本发明涉及一种化学中间体的制备方法,尤其是涉及一种高品2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶的合成方法。
背景技术:
2.kras g12c抑制剂是一种用于治疗胰腺癌、结肠直肠癌和肺癌的药物,而2
‑
异丙基
‑4‑
甲基吡啶
‑3‑
胺是制备kras g12c抑制剂的重要中间体。
3.关于2
‑
异丙基
‑4‑
甲基吡啶
‑3‑
胺的制备方法报道较少,现在已知的有明确报道的是wo2020/81636、us2019/374542和cn111377918,这些方法以3
‑
氨基
‑4‑
甲基吡啶为原料,以nbs/三氟醋酸为溴代试剂,合成3
‑
氨基
‑2‑
溴
‑4‑
甲基吡啶,然后与异丙基溴化锌/异丙基氯化镁进行偶联合成2
‑
异丙基
‑4‑
甲基吡啶
‑3‑
胺;此外,cn112159405以异丙烯基硼酸频哪醇酯与3
‑
硝基
‑2‑
氯
‑4‑
甲基吡啶进行偶联,然后用10%的氢氧化钯进行还原合成2
‑
异丙基
‑4‑
甲基吡啶
‑3‑
胺等类似反应。
4.对以上方案进行分析,发现以上方法中,nbs/三氟醋酸溴代时,虽然可得到产品3
‑
氨基
‑2‑
溴
‑4‑
甲基吡啶,但是成本较高,产品3
‑
氨基
‑2‑
溴
‑4‑
甲基吡啶收率偏低。而异丙基溴化锌/异丙基氯化镁成本高,格氏试剂活性高,具有安全隐患。用异丙烯基硼酸频哪醇酯与3
‑
硝基
‑2‑
氯
‑4‑
甲基吡啶进行偶联,原料成本偏高,而且需要用到贵金属催化剂,用量较大,成本太高,不利于工业化生产。
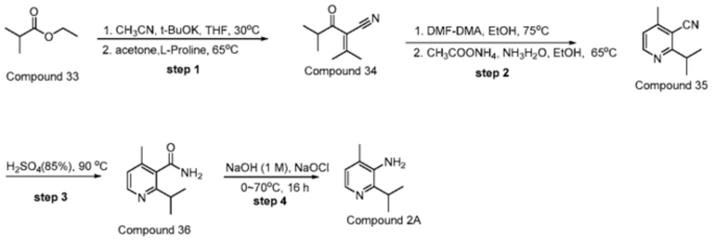
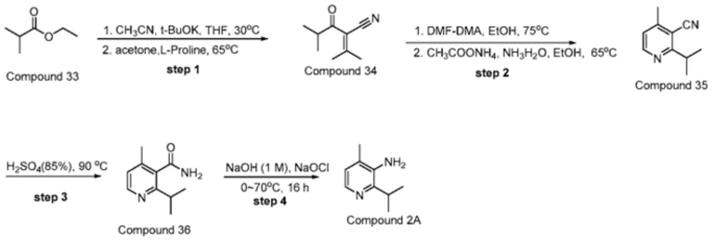
5.wo2021097207里面介绍了以异丁酸乙酯为起始原料的合成方法,合成路线如下:
[0006][0007]
该方法所用的原料和试剂比较便宜易得,但是所有步骤都没有给出后处理方式、反应收率和纯度等参数,因此难以对其效果进行评价,申请人重复了对其试验过程,结果发现该方法存在以下未说明的缺陷:(1)从compound 33(异丁酸乙酯)到compound 34(2
‑
异丁酰基
‑3‑
甲基
‑2‑
丁烯腈)的步骤,第二步是用l
‑
脯氨酸作为催化剂的,但是本发明人发现针对该底物,l
‑
脯氨酸的催化效果极差,需要加入一定量的分子筛反应才会发生,并且收率较低,详见对比实施例1和对比例实施例2;(2)该路线中compound 35(2
‑
异丙基
‑4‑
甲基
‑3‑
氰基吡啶)需要经过硫酸酸解,中和处理出来再进行hofmann降解得到2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶这一步骤中。首先85%的硫酸在90℃下酸解的过程中反应液颜色变黑,有部分中间体被硫酸碳化,收率低,详见对比实施例3;其次在生产上中和硫酸会产生大量的废水,对环境不友好。
技术实现要素:
[0008]
本发明提供了一种2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶的合成方法,在提高产品品质和收率的同时减少环境污染,整个合成方法反应简便、环保,产品更适合大批量生产。
[0009]
一种2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶的合成方法,包括以下步骤:
[0010]
(1)异丁酸乙酯与乙腈进行一次缩合反应,得到的缩合中间体与丙酮在l
‑
丙氨酸和分子筛的条件下二次缩合得到2
‑
异丁酰基
‑3‑
甲基
‑2‑
丁烯腈;
[0011]2‑
异丁酰基
‑3‑
甲基
‑2‑
丁烯腈的结构式如下:
[0012][0013]
(2)2
‑
异丁酰基
‑3‑
甲基
‑2‑
丁烯腈依次经过甲叉化缩合和氨吡啶环化得到2
‑
异丙基
‑4‑
甲基
‑3‑
氰基吡啶;
[0014]2‑
异丙基
‑4‑
甲基
‑3‑
氰基吡啶的结构如下:
[0015][0016]
(3)2
‑
异丙基
‑4‑
甲基
‑3‑
氰基吡啶在碱的作用下依次进行水解和hofmann降解得到2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶。
[0017]
作为优选,步骤(1)中,一次缩合反应在thf中进行。
[0018]
作为优选,步骤(1)中,缩合反应在叔丁醇钾中进行。
[0019]
步骤(1)中,乙腈的用量为过量,一次缩合反应结束之后,用盐酸调ph至3
‑
4,过滤,有机相旋干得到粗品,然后直接进行二次缩合反应。
[0020]
步骤(2)中,所述的甲叉化缩合和氨吡啶环化可参考现有技术的方法进行操作。
[0021]
步骤(2)中,氨吡啶环化的n源,可选自氨水、氨甲醇溶液、氨乙醇溶液或者醋酸铵,优选为醋酸铵。
[0022]
作为优选,步骤(3)的水解和hofmann降解可以分步进行或者以一锅法的方式进行。
[0023]
作为优选,步骤(3)中,所述的碱为氢氧化钠、氢氧化锂或者氢氧化钾,进一步优选为氢氧化钠。
[0024]
作为优选,当反应以分步方式进行的时候,所述的水解在醇溶剂和碱中进行;
[0025]
水解结束后蒸干溶剂,水洗,然后加入碱和水,再滴加溴或次氯酸钠进行hofmann降解,得到2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶。
[0026]
作为另外的优选,当反应以一锅法的方式进行的时候,所述的水解在醇溶剂和碱中进行,反应结束之后,不进行后处理直接加水,然后滴加溴或次氯酸钠进行hofmann降解,得到2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶。
[0027]
步骤(3)中,作为优选,所述水解反应的温度为醇溶剂的回流温度。
[0028]
步骤(3)中,作为优选,所述的hofmann降解中,溴或次氯酸钠在5℃以下滴加,滴加完成之后,现在15~30℃下反应2~3h,然后加热至65~75℃继续反应。
[0029]
步骤(3)中,作为优选,反应结束之后,所述的2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶采用石油醚打浆进行提纯。
[0030]
步骤(3)中,作为优选,所述的2
‑
异丙基
‑4‑
甲基
‑3‑
氰基吡啶、次氯酸钠、碱的摩尔比为1:(1.0
‑
2.0):(3.0
‑
6.0)。
[0031]
步骤(3)中,作为优选,所述的醇溶剂为异丙醇。
[0032]
本发明的具体合成路线如下:
[0033][0034]
同现有技术相比,本发明的有益效果体现在:
[0035]
(1)本发明的方法在提高产品质量品质的同时减少环境污染,使产品生产清洁化、环保化,使产品更适合大批量生产。
[0036]
(2)本发明采用l
‑
丙氨酸代替l
‑
脯氨酸进行缩合反应,意外地发现反应收率大幅度提高;
[0037]
(3)本发明的方法采用氢氧化钠进行水解,与酸解方法相比,不需要进行中和即可直接进入后一步hofmann降解,操作更简单,对环境更加友好;
[0038]
(4)由于水解和hofmann降解都在碱性条件下反应,因而氰基转化为氨基可以采用一锅法,绿色环保,工业化生产和成本降低。
具体实施方式
[0039]
下面结合具体实施例对本发明作进一步说明,但本发明的保护范围并不仅限于此。
[0040]
实施例1
[0041][0042]2‑
异丁酰基
‑3‑
甲基
‑2‑
丁烯腈合成:在装有温度计、搅拌器的四口烧瓶中,加入11.6g异丁酸乙酯,60mlthf及20g叔丁醇钾,开启搅拌,控制反应温度45~50℃,滴加70g乙腈,保温反应4h,原料反应完毕后,降温至10℃以下,然后加入冰盐酸调ph至3
‑
4,过滤,有机相旋干得到粗品11.1g。
[0043]
在装有温度计、搅拌器四口烧瓶中,加入11.1g上述所得粗品,80ml丙酮,100g 4a分子筛及1.11g l
‑
丙氨酸,开启搅拌,升温至回流进行反应,保温反应4h,原料反应完毕后,过滤回收4a分子筛,有机相蒸干回收丙酮,然后加入30ml二氯甲烷溶解,再加30ml水洗、分液,旋干回收二氯甲烷得到2
‑
异丁酰基
‑3‑
甲基
‑2‑
丁烯腈12.6g,纯度95.6%,收率83.4%。
[0044]
对比例1
[0045][0046]2‑
异丁酰基
‑3‑
甲基
‑2‑
丁烯腈合成:在装有温度计、搅拌器四口烧瓶中,加入11.6g异丁酸乙酯,60mlthf及20g叔丁醇钾,开启搅拌,控制反应温度45~50℃,滴加70g乙腈,保温反应4h,原料反应完毕后,降温至10℃以下,然后加入冰盐酸调ph至3
‑
4,过滤,有机相旋干得到粗品。
[0047]
在装有温度计、搅拌器四口烧瓶中,加入该粗品,80ml丙酮,1.11g l
‑
脯氨酸,开启搅拌,升温至回流进行反应,保温反应8h,几乎未反应。
[0048]
从对比例1中可以看出wo2021097207中l
‑
脯氨酸催化反应效果很差。
[0049]
对比例2
[0050][0051]2‑
异丁酰基
‑3‑
甲基
‑2‑
丁烯腈合成:在装有温度计、搅拌器四口烧瓶中,加入11.6g异丁酸乙酯,60mlthf及20g叔丁醇钾,开启搅拌,控制反应温度45~50℃,滴加70g乙腈,保温反应4h,原料反应完毕后,降温至10℃以下,然后加入冰盐酸调ph至3
‑
4,过滤,有机相旋干得到粗品。
[0052]
在装有温度计、搅拌器四口烧瓶中,加入11.1g该粗品,80ml丙酮,100g 4a分子筛及1.11g l
‑
脯氨酸,开启搅拌,升温至回流进行反应,保温反应14h,gc显示原料约剩余65%,停止反应,未进行后处理。
[0053]
该对比例可以看出wo2021097207中l
‑
脯氨酸有分子筛存在的条件下催化效果也不好。
[0054]
实施例2
[0055][0056]5‑
n,n
‑
二甲基
‑2‑
异丁酰基
‑
2,4
‑
二戊烯腈的合成:在装有温度计、搅拌器四口烧瓶中,加入15.1g 2
‑
异丁酰基
‑3‑
甲基
‑2‑
丁烯腈,60ml乙醇及11.9g dmf
‑
dma,开启搅拌,升温至回流反应,保温反应3h,原料反应完毕后,蒸干溶剂,加入50ml水打浆,过滤,烘干得到5
‑
n,n
‑
二甲基
‑2‑
异丁酰基
‑
2,4
‑
二戊烯腈19.1g,纯度99.5%,收率93%。
[0057]
实施例3
[0058][0059]2‑
异丙基
‑4‑
甲基
‑3‑
氰基吡啶的合成:在装有温度计、搅拌器四口烧瓶中,加入20.6g 5
‑
n,n
‑
二甲基
‑2‑
异丁酰基
‑
2,4
‑
二戊烯腈,80ml乙醇及30.8g醋酸铵,开启搅拌,升温至回流反应,保温反应6h,原料反应完毕后,蒸干溶剂,加入50ml水洗,并用二氯甲烷萃取,分液,旋干回收二氯甲烷得到2
‑
异丙基
‑4‑
甲基
‑3‑
氰基吡啶12.5g,纯度99.3%,收率78%。
[0060]
实施例4
[0061][0062]2‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶合成:在装有温度计、搅拌器四口烧瓶中,加入52.6g 2
‑
异丙基
‑4‑
甲基
‑3‑
氰基吡啶,150ml异丙醇及66g氢氧化钠,开启搅拌,升温至回流反应,保温反应12h,原料反应完毕后,加入33.7g水,降温至0℃以下,开始滴加111.0g次氯酸钠水溶液(7.5%含量)控温在5℃以下,保温15min,转至室温下,反应2h,后加热至70℃反应1h,分析无中间态为止,停止反应,蒸出异丙醇,残液中加入浓盐酸调ph至2~3,用53ml二氯甲烷萃取分液,水相用氢氧化钠固体调ph至11~12,然后用100nl甲苯萃取,旋干有机相,得粗品,然后加入50ml石油醚打浆,过滤即可得2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶38.6g,hplc纯度99.7%,收率78.2%。
[0063]
产物表征数据如下:1hnmr(500mhz,cdcl3):1.31(d,6h);2.31(s,3h);3.44(m,1h);5.79(s,2h);7.35(d,1h);8.09(d,1h)。
[0064]
实施列5
[0065][0066]2‑
异丙基
‑4‑
甲基烟酰胺合成:在装有温度计、搅拌器四口烧瓶中,加入16g 2
‑
异丙基
‑4‑
甲基
‑3‑
氰基吡啶,80ml异丙醇及16g氢氧化钠,开启搅拌,升温至回流反应,保温反应12h,原料反应完毕后,蒸干溶剂,加入50ml水洗,过滤得到2
‑
异丙基
‑4‑
甲基烟酰胺15g,纯度98.1%,收率84%。
[0067]
对比例3
[0068][0069]2‑
异丙基
‑4‑
甲基烟酰胺合成:在装有温度计、搅拌器四口烧瓶中,加入70.5g 85%硫酸,开启搅拌,分批加入16g 2
‑
异丙基
‑4‑
甲基
‑3‑
氰基吡啶,加完后升温至90℃,保
温反应16h,原料反应完毕后,反应液颜色变深有部分碳化,降温到0℃,并滴加饱和碳酸钠水溶液调反应液的ph到8,加入二氯甲烷萃取反应液,蒸干溶剂用16ml异丙醇与48ml石油醚重结晶滤饼,过滤得到2
‑
异丙基
‑4‑
甲基烟酰胺(6.2g),纯度98.4%,收率34.7%,由此可见,当采用硫酸时有部分中间体被硫酸碳化,收率偏低。
[0070]
实施例6
[0071][0072]2‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶的合成:
[0073]
在装有温度计、搅拌器四口烧瓶中,加入50g 2
‑
异丙基
‑4‑
甲基烟酰胺,33.7g氢氧化钠与183.7g水,开启搅拌,降温至0℃以下,开始滴加111g次氯酸钠水溶液(7.5%含量)控温在15℃以下反应2h,后加热至70℃反应完全为止,停止反应,加入浓盐酸调ph至2
‑
3,用50ml二氯甲烷萃取杂质,分液,水相用氢氧化钠固体调ph至11
‑
12,然后用2倍甲苯萃取,旋干有机相,然后加入正庚烷打浆,过滤即可得到2
‑
异丙基
‑3‑
氨基
‑4‑
甲基吡啶38g,纯度99.6%,收率90%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。