1.本发明属于医药领域,具体涉及一种桥环嘧啶酮类化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式、其制备方法、其组合物、以及其在医药上的用途。特别地,本公开涉及通式(i)所示的桥环嘧啶酮类衍生物、其制备方法及含有该衍生物的药物组合物,以及其作为lppla2抑制剂治疗阿尔茨海默病、青光眼、年龄相关性黄斑变性(amd)等神经退行性疾病或动脉粥样硬化、糖尿病性黄斑水肿的用途。
背景技术:
2.脂蛋白相关磷脂酶a2(lp-pla2)是磷脂酶a2超家族的成员(dennis ea,cao j,hsu yh,magrioti v,kokotos g.chem rev.2011,111,6130-6185)。它主要由单核细胞,巨噬细胞,t淋巴细胞和主细胞分泌(stafforini dm,elstad mr,mcintyre tm,zimmerman ga,prescott sm.j biol chem.1990,265:9682-9687;nakajima k,murakami m,yanoshita r,samejima y,karasawa k,setaka m,nojima s kudo i.j biol chem.1997,272,19708-19713)。磷脂酰胆碱sn-2酯是在低密度脂蛋白(ldl)氧化过程中产生的,lp-pla2负责水解氧化修饰的磷脂酰胆碱sn-2酯,然后产生氧化脂肪酸和溶血磷脂酰胆碱(lysopc)(caslake mj,packard cj,suckling ke,holmes sd,chamberlain p,macphee ch.atherosclerosis.2000,150,413-419;macphee ch,moores ke,boyd hf,dhanak d,ife rj,leach ca,leake ds,milliner kj,patterson ra,suckling ke,tew dg,hickey dm.biochem j.1999,338,479-487)。氧化脂肪酸和lysopc都在活化巨噬细胞、增加氧化应激、影响t淋巴细胞功能和诱导炎症反应中发挥作用(quinn mt,parthasarathy s,steinberg d.proc natl acad sci u s a.1988,85,2805-2809)。据报道,lysopc诱导多种细胞毒性炎性细胞因子释放(shi,et al,atherosclerosis,2007,191,54-62)。此外,lysopc还涉及白细胞活化,细胞凋亡的诱导和内皮功能障碍的介导(wilensky et al,current opinion in lipidology,2009,20,415-420)。
3.有文献报道,血浆的lp-pla2水平与心血管疾病(fitzpatrick al,irizarry mc,cushman m,jenny ns,chi gc,koro c.atherosclerosis.2014,235,384-391),糖尿病性黄斑水肿(dme)(staurenghi g,ye l,magee mh,danis rp,wurzelmann j,adamson p,mclaughlin mm,darapladib dmesg.ophthalmology.2015,122,990-996),前列腺癌(bertilsson h,tessem mb,flatberg a,viset t,gribbestad i,angelsen a,halgunset j.clin cancer res.2012,18,3261-3269)相关。
4.阿尔茨海默病(ad)是一种慢性神经退行性疾病,它会导致认知能力下降,情绪波动,不可逆转的记忆丧失,方向障碍,语言障碍和失去自我保护能力(hardy j,et al.science2002,297,353-356.)。阿尔茨海默病通常随着时间的推移开始缓慢并逐渐恶化,这是60%至70%的痴呆病例的原因,并影响65岁以上人口的约6%。ad患者将逐渐退出家庭和社会,越来越依赖帮助,最终进展至死亡。ad是发达国家中代价最高的疾病之一,在其他国家的成本也很高。特别是随着老龄化成为一个重要的社会问题,这些成本将急剧增
加。毋庸置疑,ad是一种复杂的涉及多种因素的疾病。尽管ad的病因尚未完全阐明,但很明显,有几个因素参与疾病的发生和发展,包括聚集的tau蛋白和aβ肽,氧化应激和神经炎症(echeverria v,yarkov a,aliev g.prog neurobiol.2016,144,142-157)。目前的ad药物研发主要集中在aβ淀粉样变性和tau的靶点上(chiang k,koo eh.annu rev pharmacol toxicol.2014,54,381-405;awasthi m,singh s,pandey vp,dwivedi un.j neurol sci.2016,361,256-271)。然而,尽管临床前数据很强,但后期临床试验的结果迄今未能证明临床疗效。这些令人失望的结果预示,针对ad治疗可能必须探索其他神经病理学机制,例如氧化应激和神经炎症。
5.血浆中lp-pla2水平的升高增加了患痴呆症的风险,包括ad(van oijen,et al.annals of neurology,2006,59,139),ad患者中发现了血管性痴呆和混合性痴呆,以及高氧化ldl水平(maher-edwards g,de’ath j,barnett c,lavrov a,lockhart a,alzheimer’s&dementia:translational research&clinical interventions.2015,1,131-140;kassner et al.current alzheimer research,2008,5,358-366;dildar,et al.,alzheimer dis assoc disord,24,april
–
june(2010);sinem,et al.current alzheimer research,2010,7,463-469)。在ad患者中也发现了神经炎症和上调的多种炎性细胞因子(colangelo,et al.,journal of neuroscience research,2002,70,462
–
473;wyss-coray,nature medicine,2006,12,sept.)。
6.基于所有这些发现,lp-pla2是治疗ad的潜在靶点,而lp-pla2抑制剂rilapladib针对ad患者的临床结果进一步证实了这一点(maher-edwards g,de’ath j,barnett c,lavrov a,lockhart a,alzheimer’s&dementia:translational research&clinical interventions.2015,1,131-140)。
7.青光眼和年龄相关性黄斑变性(amd)属于视网膜神经退行性疾病。buschini等报道炎症包括tnf-α信号,可能在青光眼和amd的发病机制中起重要作用(buschini et al,progress in neurobiology,2011,95,14-25;tezel,progress in brain research,vol.173,issn0079-6123,chapter 28)。另外,shi等人证实lp-pla2抑制剂可以阻断炎性细胞因子的释放(shi,et al,atherosclerosis,2007,191,54-62)。lp-pla2的抑制是青光眼和amd的潜在治疗方法。
8.已经报道了许多lp-pla2抑制剂,包括β-内酰胺(tew dg,boyd hf,ashman s,theobald c,leach ca.biochemistry.1998,37,10087-10093),肟(jeong ts,kim mj,yu h,kim hs,choi jk,kim ss,lee ws.bioorg med chem lett.2005,15,1525-1527;jeong hj,park yd,park hy,jeong iy,jeong ts,lee ws.bioorg med chem lett.2006,16,5576-5579),黄尿酸的酰胺(lin ec,hu y,amantea cm,pham lm,cajica j,okerberg e,brown he,fraser a,du l,kohno y,ishiyama j,kozarich jw,shreder kr.bioorg med chem lett.2012,22,868-871;hu y,lin ec,pham lm,cajica j,amantea cm,okerberg e,brown he,fraser a,du l,kohno y,ishiyama j,kozarich jw,shreder kr.bioorg med chem lett.2013,23,1553-1556.),和氨基甲酸酯(nagano jm,hsu kl,whitby lr,niphakis mj,speers ae,brown sj,spicer t,fernandez-vega v,ferguson j,hodder p,srinivasan p,gonzalez td,rosen h,bahnson bj,cravatt bf.bioorg med chem lett.2013,23,839-843)。
9.据报道lp-pla2抑制剂darapladib是一种抗动脉粥样硬化和dme的潜在治疗方法(magrioti v,kokotos g.expert opin ther pat.2013;23:333-344)。
技术实现要素:
10.本发明人发现,lp-pla2抑制剂在治疗神经退行性相关疾病,如阿尔茨海默病(ad)、青光眼和年龄相关的黄斑变性(amd),或者包括动脉粥样硬化等的心血管疾病方面有着重要的作用。为此,本发明人致力于开发出一种全新的lp-pla2抑制剂,桥环嘧啶酮类化合物。
11.该桥环嘧啶酮类化合物为具有式(i)所示结构的化合物或其药学上可接受的盐,
[0012][0013]
其中,
[0014]
n1、n2、n3各自独立地为0、1或2;
[0015]
r1、r2各自独立地选自:-h、羟基、氰基、卤素、烷基、氘代烷基、氘代烷氧基、羟烷基、卤代烷基、卤代烷氧基、环烷基、烷氧基、亚芳基或亚杂芳基;
[0016]
x1、x2各自独立地选自:亚烷基、-o-、-s-或-nr'-,
[0017]
r'选自:-h、烷基、氘代烷基或环烷基;
[0018]
ar为亚芳基或亚杂芳基;所述亚芳基或亚杂芳基中的氢原子任选被一个或多个取代基取代,所述取代基各自独立地选自:卤素、烷基、氘代烷基、卤代烷基、烷氧基、氘代烷氧基、卤代烷氧基、羟基、羟烷基、氰基、氨基、单烷基或二烷基取代的氨基、硝基、羧基、醛基、环烷基、杂环基、芳基或杂芳基;
[0019]
y为-h、卤素、烷基、卤代烷基、卤代烷氧基、环烷基、烷氧基、氘代烷基、氘代烷氧基、羟基、羟烷基、氰基、亚芳基、亚杂芳基、-oar'、-sar'、-nr
”-
ar'、-n r”r”或-r”'-ar';
[0020]
ar'选自芳基或杂芳基;所述芳基或杂芳基中的氢原子任选被一个或多个取代基取代,所述取代基各自独立地选自:卤素、烷基、卤代烷基、烷氧基、羟基、羟烷基、卤代烷氧基、氘代烷基、氘代烷氧基、氰基、氨基、硝基、羧基、醛基、环烷基、杂环基、芳基或杂芳基;
[0021]
r”为h、烷基或环烷基;
[0022]
r”'为亚烷基;
[0023]
z为o或s。
[0024]
可选地,所述“卤素”、“卤代烷基”、“卤代烷氧基”中所具有的卤原子各自独立地选自f、cl、br或i;
[0025]
可选地,所述“烷基”、“氘代烷基”、“氘代烷氧基”、“羟烷基”、“卤代烷基”、“卤代烷氧基”、“烷氧基”、“单烷基或二烷基取代的氨基”中所具有的烷基各自独立地为c
1-c
10
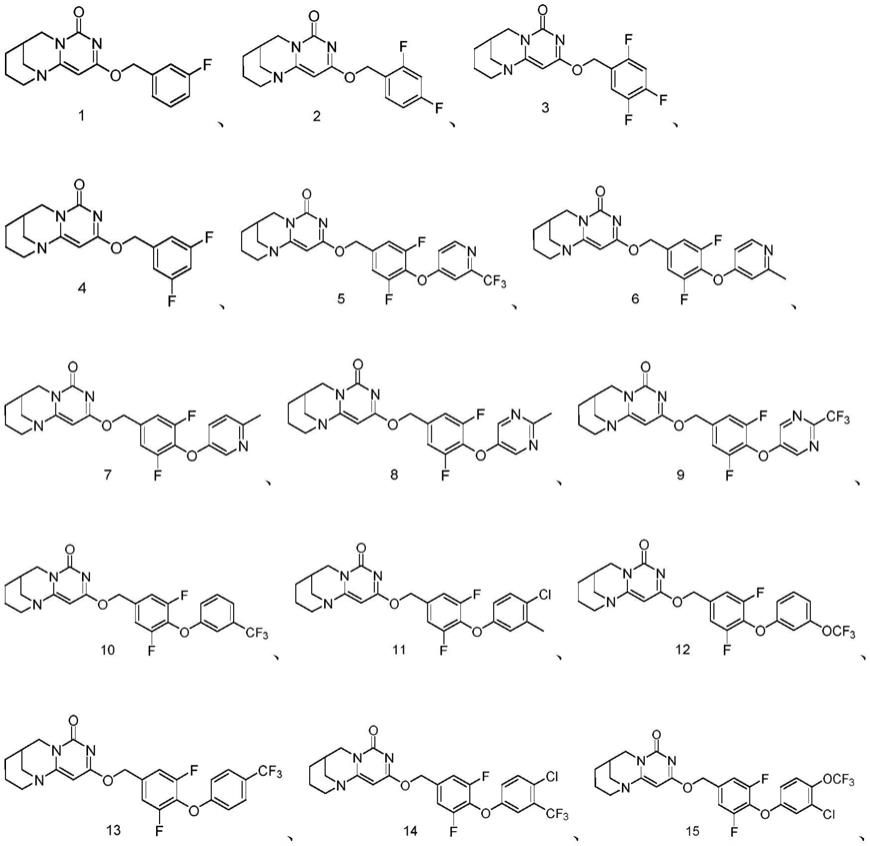
直链或支链烷基;可选地,为c
1-c7直链或支链烷基;可选地,为c
1-c4直链或支链烷基;可选地,选自甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基,1-甲基丁基、2-甲
基丁基、3-甲基丁基、异戊基、1-乙基丙基、新戊基、正己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、异己基、1,1-二甲基丁基、2,2-二甲基丁基、3,3-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,3-二甲基丁基、2-乙基丁基、正庚基、2-甲基己基、3-甲基己基、2,2-二甲基戊基、3,3-二甲基戊基、2,3-二甲基戊基、2,4-二甲基戊基、3-乙基戊基或2,2,3-三甲基丁基;
[0026]
可选地,所述“亚烷基”各自独立地为c
1-c
10
的直链或支链的亚烷基;可选地,为c
1-c7直链或支链亚烷基;可选地,为c
1-c5直链或支链亚烷基;可选地,选自亚甲基、亚乙基、亚正丙基、亚异丙基、亚正丁基、亚异丁基、亚叔丁基、亚仲丁基、亚正戊基,1-甲基亚丁基、2-甲基亚丁基、3-甲基亚丁基、亚异戊基、1-乙基亚丙基、亚新戊基、亚正己基、1-甲基亚戊基、2-甲基亚戊基、3-甲基亚戊基、亚异己基、1,1-二甲基亚丁基、2,2-二甲基亚丁基、3,3-二甲基亚丁基、1,2-二甲基亚丁基、1,3-二甲基亚丁基、2,3-二甲基亚丁基、2-乙基亚丁基、亚正庚基、2-甲基亚己基、3-甲基亚己基、2,2-二甲基亚戊基、3,3-二甲基亚戊基、2,3-二甲基亚戊基、2,4-二甲基亚戊基、3-乙基亚戊基或2,2,3-三甲基亚丁基;
[0027]
可选地,所述“环烷基”为c
3-c
10
单环或双环环烷基,可选地,为c
3-c7单环环烷基,可选地,为环丙基、环丁基、环戊基、环已基或环庚基;
[0028]
可选地,所述“杂环基”为环上含有1个、2个或3个选自n、o、s的杂原子的3-10元非芳香杂环,可选地,所述杂环为环上含有1个或2个选自n、o的杂原子的3-10元非芳香环;可选地,所述杂环为环上含有1个或2个选自n、o的杂原子的3-6元非芳香环;可选地,所述杂环为环上含有1个或2个选自n、s的杂原子的3-10元非芳香环;可选地,所述杂环为环上含有1个或2个选自n、s的杂原子的3-6元非芳香环;
[0029]
可选地,所述“芳基”为6-10元芳基;可选为苯基或萘基,可选为苯基、1-萘基、或2-萘基;
[0030]
可选地,所述“亚芳基”为6-10元亚芳基;可选为亚苯基或亚萘基;
[0031]
可选地,所述“杂芳基”为环上含有1-3个选自n、o和s中的杂原子的5-10元杂芳环;可选地,为环上含有1-2个选自n、o和s中的杂原子的5-10元杂芳环;可选地,所述杂芳环选自吡啶环、吡咯环、吡唑环、嘧啶环、吡嗪环、哒嗪环、噻吩环、呋喃环;可选地,所述杂芳基选自吡啶-2-基、吡啶-3-基、吡啶-4-基、哒嗪-3-基、哒嗪-4-基、嘧啶-2-基、嘧啶-4-基、嘧啶-5-基、吡嗪-2-基、吡嗪-3-基、吲哚基、异吲哚基、吲唑基、吲嗪基、嘌呤基、喹嗪基、喹啉基、异喹啉基、噌啉基、酞嗪基、萘啶基、喹唑啉基、喹喔啉基、噻吩并[2,3-b]呋喃基、呋喃并[3,2-b]-吡喃基、吡啶并[2,3-d]噁嗪基、吡唑并[4,3-d]噁唑基、咪唑并[4,5-d]噻唑基、吡嗪并[2,3-d]哒嗪基、咪唑并[2,1-b]噻唑基、咪唑并[1,2-b][l,2,4]三嗪基、苯并噻吩基、苯并噁唑基、苯并咪唑基、苯并噻唑基、苯并噁庚因基、苯并噁嗪基、苯并呋喃基、苯并三唑基、吡咯并[2,3-b]吡啶基、吡咯并[3,2-c]吡啶基、吡咯并[3,2-b]吡啶基、咪唑并[4,5-b]吡啶基、咪唑并[4,5-c]吡啶基、吡唑并[4,3-d]吡啶基、吡唑并[4,3-c]吡啶基、吡唑并[3,4-c]吡啶基、吡唑并[3,4-d]吡啶基、吡唑并[3,4-b]吡啶基、咪唑并[1,2-a]吡啶基、吡唑并[1,5-a]吡啶基、吡咯并[1,2-b]哒嗪基、咪唑并[1,2-c]嘧啶基、吡啶并[3,2-d]嘧啶基、吡啶并[4,3-d]嘧啶基、吡啶并[3,4-d]嘧啶基、吡啶并[2,3-d]嘧啶基、吡啶并[2,3-b]吡嗪基、吡啶并[3,4-b]吡嗪基、嘧啶并[5,4-d]嘧啶基、吡唑并[2,3-b]吡嗪基、或嘧啶并[4,5-d]嘧啶基,可选为吡啶-2-基、吡啶-3-基、吡啶-4-基、嘧啶-2-基、嘧啶-4-基或嘧啶-5-基;
[0032]
可选地,所述“亚杂芳基”为环上含有1-3个选自n、o和s中的杂原子的5-10元亚杂芳环;可选地,为环上含有1-2个选自n、o和s中的杂原子的5-10元亚杂芳环;可选地,所述亚杂芳环选自亚吡啶环、亚吡咯环、亚吡唑环、亚嘧啶环、亚吡嗪环、亚哒嗪环、亚噻吩环、或亚呋喃环。
[0033]
可选地,所述式(i)所示的化合物为其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或这些异构体混合物形式。
[0034]
可选地,n1、n2、n3各自独立地为0、1或2。
[0035]
可选地,n1为1。
[0036]
可选地,n2为1。
[0037]
可选地,n3为1。
[0038]
可选地,r1、r2各自独立地选自:-h、氟、氯、溴、碘、羟基、羟烷基、氰基、c
1-c7烷基(如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基,1-甲基丁基、2-甲基丁基、3-甲基丁基、异戊基、1-乙基丙基、新戊基、正己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、异己基、1,1-二甲基丁基、2,2-二甲基丁基、3,3-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,3-二甲基丁基、2-乙基丁基、正庚基、2-甲基己基、3-甲基己基、2,2-二甲基戊基、3,3-二甲基戊基、2,3-二甲基戊基、2,4-二甲基戊基、3-乙基戊基或2,2,3-三甲基丁基)、c
1-c3氘代烷基(如-cd3、-c2d5、或-c3d7)、c
1-c3氘代烷氧基(如-ocd3、-oc2d5、或-oc3d7)、c
1-c3卤代烷基(如-cf3、-chf2、-ch2f、-c2f5、-c3f7)、c
1-c7卤代烷氧基、c
1-c7烷氧基、环丙烷基、环丁烷基、环戊烷基、或环己烷基;可选地,r1为-h;可选地,r2为-h;
[0039]
可选地,x1、x2各自独立地选自:c
1-c7亚烷基、-o-、-s-或-nr'-;可选地,x1为c
1-c7亚烷基(可选为,-ch
2-,亚乙基、亚正丙基、亚异丙基、亚正丁基或亚异丁基)、-o-、或-s-;可选地,x1为c
1-c7亚烷基或-o-;可选地,x1为-ch
2-或-o-;可选地,x2为-o-或-s-;可选地,x2为-o-;
[0040]
可选地,r'选自-h、c
1-c7烷基(如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基,1-甲基丁基、2-甲基丁基、3-甲基丁基、异戊基、1-乙基丙基、新戊基、正己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、异己基、1,1-二甲基丁基、2,2-二甲基丁基、3,3-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,3-二甲基丁基、2-乙基丁基、正庚基、2-甲基己基、3-甲基己基、2,2-二甲基戊基、3,3-二甲基戊基、2,3-二甲基戊基、2,4-二甲基戊基、3-乙基戊基或2,2,3-三甲基丁基)、氘代烷基(可选为-cd3、-c2d5、或-c3d7)或c
3-c6环烷基(如环丙烷基、环丁烷基、环戊烷基、或环己烷基);
[0041]
可选地,ar为亚苯基或吡啶基,所述亚苯基或吡啶基中的氢原子任选被1、2或3个取代基取代,所述取代基各自独立地选自:f、cl、br、i、-cn、-me、-cf3、-chf2、-c2h5、-c3h7、环丙基、环丁基、环戊基、环己基、-cd3、-ocd3、-ome、-ocf3、或-ochf2;
[0042]
可选地,ar为亚芳基;可选地,ar为亚苯基,所述亚苯基中的氢原子任选被1个或2个取代基取代,所述取代基为卤素;可选地,所述取代基为f;
[0043]
可选地,y为-h、-f、-cl、-br、-i、甲基、乙基、正丙基、异丙基、-cd3、-ocd3、-cf3、-chf2、-ch2f、-ch2cf3、-ocf3、-ochf2、-och2f、环丙基、-环丁基、-环戊基、环己基、-och3、-oc2h5、-oc3h7、或-oar';
[0044]
可选地,y为h、卤素、或-oar';可选地,y为h、-f、或-oar';
[0045]
可选地,ar'选自苯基、吡啶基、嘧啶基、噻吩基、吡咯基、吡唑基、或喹啉基,所述苯基、吡啶基、嘧啶基、噻吩基、吡咯基、吡唑基、或喹啉基环中的氢原子各自独立地任选被1、2或3个取代基取代,所述取代基各自独立地选自:f、cl、br、-cn、c
1-c7烷基(可选为甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基,1-甲基丁基、2-甲基丁基、3-甲基丁基、异戊基、1-乙基丙基、新戊基、正己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、异己基、1,1-二甲基丁基、2,2-二甲基丁基、3,3-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,3-二甲基丁基、2-乙基丁基、正庚基、2-甲基己基、3-甲基己基、2,2-二甲基戊基、3,3-二甲基戊基、2,3-二甲基戊基、2,4-二甲基戊基、3-乙基戊基或2,2,3-三甲基丁基)、-cd3、-ocd3、c
1-c6卤代烷基、-och3、-oc2h7、-oc3h7、c
1-c6卤代烷氧基、羟基、羟烷基、氰基、或c
3-c6环烷基(可选为环丙烷基、环丁烷基、环戊烷基、环己烷基);
[0046]
可选地,ar'选自苯基、吡啶-3-基、吡啶-4-基或嘧啶-5-基,可选地,所述ar'被1个或2个取代基取代,所述取代基选自卤素、烷基、卤代烷基或卤代烷氧基;可选地,所述取代基选自f、cl、-ch3、-cf3或-ocf3;
[0047]
可选地,z为o或s;可选地,z为o。
[0048]
可选地,所述式(i)化合物,或其药学上可接受的盐中,所述式(i)化合物选自下列化合物:
[0049]
[0050][0051]
可选地,所述的式(i)化合物或其药学上可接受的盐,其特征在于,所述药学上可接受的盐包括式(i)化合物的阴离子盐或阳离子盐;
[0052]
可选地,所述药学上可接受的盐包括式i化合物的碱金属的盐、碱土金属的盐、或铵盐;可选地,所述碱金属包括钠、钾、锂、或铯,所述碱土金属包括镁、钙、或锶;
[0053]
可选地,所述药学上可接受的盐包括式i化合物与有机碱形成的盐;
[0054]
可选地,所述有机碱包括三烷基胺、吡啶、喹啉、哌啶、咪唑、甲基吡啶、二甲氨基吡啶、二甲基苯胺、n-烷基吗啉、1,5-二氮杂双环[4.3.0]壬烯-5、1,8-二氮杂双环[5.4.0]十一碳烯-7、1,4-二氮杂双环[2.2.2]辛烷;可选地,所述三烷基胺包括三甲胺、三乙胺、或n-乙基二异丙胺;可选地,所述n-烷基吗啉包括n-甲基吗啉;
[0055]
可选地,所述药学上可接受的盐包括式i化合物与酸形成的盐;
[0056]
可选地,所述酸包括无机酸、或有机酸;可选地,所述无机酸包括盐酸、氢溴酸、氢碘酸、硫酸、硝酸、磷酸、或碳酸;可选地,所述有机酸包括甲酸、乙酸、丙酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、乳酸、苹果酸、柠檬酸、枸橼酸、酒石酸、碳酸、苦味酸、甲磺酸、乙磺酸、对甲苯磺酸、谷氨酸、或双羟萘酸。
[0057]
另一方面,提供一种式(i)化合物或其药学上可接受的盐的制备方法,其特征在于,包括:使式(ii)化合物与式(iii)化合物反应,生成式(i)化合物的步骤:
[0058][0059]
可选地,所述制备方法包括使式(iv)化合物与三氯氧磷反应生成式(ii)化合物的步骤:
[0060][0061]
可选地,所述制备方法包括使式(v)化合物进行环化反应生成式(iv)化合物的步骤:
[0062][0063]
可选地,所述制备方法包括使式(vii)化合物与式(viii)化合物进行反应生成式(vi)化合物,并进一步由式(vi)化合物脱保护基团生成式(v)化合物的步骤:
[0064][0065]
可选地,所述制备方法包括以下反应路线:
[0066][0067]
上述制备方法中的各式中,n1、n2、n3、r1、r2、x1、x2、z、ar和y的定义如上所述。
[0068]
对上述各反应的具体反应条件并没有特别的限制,可以采用现有的常规反应条件或步骤。
[0069]
另一方面,提供一种药物组合物,其包含治疗有效量的上述式(i)化合物或其药学上可接受的盐中的一种或多种以及任选存在的药学上可接受的辅料。
[0070]
可选地,所述药物组合物的剂型包括口服制剂、直肠给药制剂、或胃肠外给药制剂;
[0071]
可选地,所述口服制剂包括固体制剂、或液体制剂;
[0072]
可选地,所述固体制剂包括片剂、粉剂、粒剂、或胶囊;
[0073]
可选地,所述液体制剂包括水或油悬浮剂、或糖浆;
[0074]
可选地,所述胃肠外给药制剂包括注射用的溶液、或者水或油性悬浮剂。
[0075]
另一方面,提供上述式(i)化合物或其药学上可接受的盐,或者上述药物组合物在制备lp-pla2抑制剂中的用途。
[0076]
另一方面,提供上述式(i)化合物或其药学上可接受的盐,或者上述药物组合物在制备治疗神经退行性相关疾病的药物中的用途;
[0077]
可选地,所述神经退行性相关疾病包括阿尔茨海默病(ad)、青光眼、年龄相关性黄斑变性(amd)。
[0078]
另一方面,提供上述的式(i)化合物或其药学上可接受的盐,或者上述药物组合物在制备治疗心血管疾病、糖尿病性黄斑水肿(dme)或前列腺疾病的药物中的用途;
[0079]
可选地,所述心血管疾病包括动脉粥样硬化。
[0080]
有益效果:
[0081]
式(i)化合物或其药学上可接受的盐是一种桥环嘧啶酮类化合物,是全新的lp-pla2抑制剂。可用于治疗神经退行性相关疾病,如阿尔茨海默病(ad)、青光眼和年龄相关的黄斑变性(amd),或者包括动脉粥样硬化等的心血管疾病、糖尿病性黄斑水肿(dme)或前列腺疾病等。
具体实施方式
[0082]
通过以下实施例对本发明进行进一步说明。应当理解的是,此处实施例仅用于示例性对本发明进行说明,并不以任何方式限制本发明的范围。
[0083]
本发明的起始原料可以采用或按照本领域已知的方法来合成,也可购买自abcr gmbh&co.kg,acros organics,aldrich chemical company,韶远化学科技(accela chembio inc)、达瑞化学品等公司。
[0084]
实施例中无特殊说明,溶液是指水溶液。
[0085]
实施例中无特殊说明,反应的温度为室温,例如20℃~30℃。
[0086]
实施例1化合物1的制备
[0087][0088][0089]
第一步:化合物1c的制备
[0090][0091]
室温下将6-氯尿嘧啶1b(8.5g,58.0mmol)、3-(羟甲基)哌啶-1-甲酸叔丁酯1a(15g,69.6mmol)和三苯基膦(22.8g,86.9mmol)溶于250ml的无水四氢呋喃和25ml的无水n,n-二甲基甲酰胺混合溶剂中,氮气保护和0℃下滴加偶氮二甲酸二异丙酯(23ml,115.8mmol),在0℃下搅拌反应2小时后,升温到室温反应过夜,过滤反应液,用乙酸乙酯萃取(50ml
×
3),合并有机相,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到无色油状产物1c(8.34g,产率:41.8%)。
[0092]
第二、三步:化合物1e的制备
[0093][0094]
室温下将化合物3-(((6-氯-2,4-二氧代-3,4-二氢嘧啶-1(2h)-基)甲基)哌啶-1-甲酸叔丁酯1c(8.34g,24.3mmol)溶于80ml的二氯甲烷中,0℃下加入20ml的三氟乙酸,室温下搅拌反应2小时,将反应液减压浓缩,直接进行下一步反应。将上一步的粗品溶于100ml乙腈中,室温下加入二异丙基乙胺(9.3g,72.9mmol),搅拌反应4小时,减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物1e(4.7g,93.3%)。
[0095]1h nmr(400mhz,dmso)δ11.01(s,1h),5.09(s,1h),3.84(m,1h),3.51(m,1h),3.30(m,1h),3.16(m,1h),3.04(m,1h),2.94(m,1h),2.25(m,1h),1.89
–
1.68(m,2h),1.61
–
1.46(m,1h),1.36(m,1h).
[0096]
第四步:化合物1f的制备
[0097][0098]
室温下将化合物1e(2.0g,9.7mmol)和二甲基苯胺(2.34g,19.3mmol)溶于甲苯中,滴加三氯氧磷(1.48g,9.7mmol),加热回流4小时后,用冰水淬灭反应,减压浓缩,用乙酸乙酯萃取(60ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物1f(0.89g,40.7%)。
[0099]1h nmr(400mhz,cdcl3)δ6.06(s,1h),4.06(m,1h),3.77(m,1h),3.52(m,1h),3.29(m,1h),3.22
–
3.15(m,1h),3.02(m,1h),2.52(m,1h),2.01
–
1.85(m,2h),1.62
–
1.44(m,2h).
[0100]
第五步:化合物1的制备
[0101][0102]
将(3-氟苯基)甲醇1g(30mg,0.24mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物1(8mg,11.5%)。
[0103]1h nmr(400mhz,cdcl3)δ7.32(m,1h),7.15(m,2h),7.04
–
6.97(m,1h),5.60(s,1h),5.40(s,2h),4.06(m,1h),3.80(m,1h),3.44(m,1h),3.29
–
3.19(m,1h),3.13(m,1h),3.03(m,1h),2.41(m,1h),2.00
–
1.82(m,2h),1.58
–
1.45(m,2h).
[0104]
实施例2化合物2的制备
[0105][0106]
将(2,4-二氟苯基)甲醇(35mg,0.24mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物2(10mg,13.6%)。
[0107]1h nmr(400mhz,cdcl3)δ7.49(m,1h),6.94
–
6.80(m,2h),5.58(s,1h),5.44(s,2h),4.09(m,1h),3.82(m,1h),3.45(m,1h),3.29
–
3.20(m,1h),3.16(m,1h),3.05(mm,1h),2.44(m,1h),2.02
–
1.83(m,2h),1.57
–
1.47(m,2h).
[0108]
实施例3化合物3的制备
[0109][0110]
将(2,4,5-三氟苯基)甲醇(39mg,0.24mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物3(12mg,15.5%)。
[0111]1h nmr(400mhz,cdcl3)δ7.32(m,1h),6.93(m,1h),5.57(s,1h),5.40(s,2h),4.06(m,1h),3.79(m,1h),3.43(m,1h),3.28
–
3.21(m,1h),3.13(m,1h),3.02(m,1h),2.42(m,1h),1.93(m,2h),1.50(m,2h).ms(esi):m/z 352.1[m h]
。
[0112]
实施例4化合物4的制备
[0113][0114]
将(3,5-二氟苯基)甲醇(35mg,0.24mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物4(6mg,8.2%)。
[0115]1h nmr(400mhz,cdcl3)δ6.99
–
6.93(m,2h),6.81
–
6.72(m,1h),5.63(s,1h),5.41(s,2h),4.09(m,1h),3.82(m,1h),3.53
–
3.43(m,1h),3.33
–
3.20(m,1h),3.17(m,1h),3.06(m,1h),2.45(m,1h),1.96(m,2h),1.54(m,2h).
[0116]
实施例5化合物5的制备
[0117][0118]
第一步:化合物5c的制备
[0119]
室温下将2-(三氟甲基)吡啶-4-醇5b(0.85g,5.2mmol),3,4,5-三氟苯甲醛5a(1g,6.2mmol)和碳酸钾(0.93g,6.76mmol)溶于30ml n,n-二甲基甲酰胺中,在90℃搅拌反应1小时,冷却到室温后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠
水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=5/1)纯化得到黄色固体产物5c(1.47g,产率:93.2%)。
[0120]1h nmr(400mhz,cdcl3)δ9.97(s,1h),8.65(m,1h),7.63(m,2h),7.27(m,1h),7.01(m,1h)。
[0121]
第二步:化合物5d的制备
[0122]
室温下将3,5-二氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苯甲醛5c(1.47g,4.85mmol)溶于50ml乙醇中,在0℃加入nabh4(184mg,4.85mmol),室温搅拌反应0.5小时,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=2/1)纯化得到白色固体产物5d(1.04g,产率:70.3%)。
[0123]1h nmr(400mhz,cdcl3)δ8.59(m,1h),7.24(m,1h),7.11(m,2h),6.99(m,1h),4.75(m,2h),2.19(m,1h)。
[0124]
第三步:化合物5的制备
[0125]
将(3,5-二氟-4
–
((2-(三氟甲基吡啶-4-基)氧基)苯基)甲醇5d(67mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物5(13mg,12%)。
[0126]1h nmr(400mhz,cdcl3)δ8.60(m,1h),7.26(s,1h),7.15(m,2h),6.99(m,1h),5.64(s,1h),5.43(s,2h),4.08(m,1h),3.80(m,1h),3.47(m,1h),3.27(m,1h),3.16(m,1h),3.04(m,1h),2.44(m,1h),1.91(m,2h),1.53(m,2h).ms(esi):m/z 494.9[m h]
。
[0127]
实施例6化合物6的制备
[0128][0129][0130]
第一步:化合物6b的制备
[0131]
室温下将2-甲基吡啶-4-醇6a(0.5g,4.6mmol),3,4,5-三氟苯甲醛5a(0.88g,5.5mmol)和碳酸钾(0.823g,5.95mmol)溶于30ml n,n-二甲基甲酰胺中,在90℃搅拌反应2小时,冷却到室温后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=10/1)纯化得到黄色固体产物6b(0.4g,产率:34.9%)。
[0132]1h nmr(400mhz,cdcl3)δ9.94(s,1h),8.39(m,1h),7.62
–
7.56(m,2h),6.70
–
6.66(m,2h),2.52(s,3h)。
[0133]
第二步:化合物6c的制备
[0134]
室温下将3,5-二氟-4-((2-甲基吡啶-4-基)氧基)苯甲醛6b(0.4g,1.6mmol)溶于50ml甲醇中,在0℃加入nabh4(71mg,1.86mmol),室温搅拌反应0.5小时,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=4/1)纯化得到无色油状产物6c(0.40g,产率:99%)。
[0135]1h nmr(400mhz,cdcl3)δ8.29(m,1h),7.07(m,2h),6.70
–
6.64(m,2h),4.73(s,2h),3.20(m,1h),2.50(s,3h)。
[0136]
第三步:化合物6的制备
[0137]
将(3,5-二氟-4
–
((2-甲基)吡啶-4-基)氧基)苯基)甲醇6c(55mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物6(8mg,8.2%)。
[0138]1h nmr(400mhz,cdcl3)δ8.36(m,1h),7.10(m,2h),6.70
–
6.66(m,2h),5.63(s,1h),5.42(s,2h),4.08(m,1h),3.81(m,1h),3.47(m,1h),3.30
–
3.22(m,1h),3.16(m,1h),3.05(m,1h),2.51(s,3h),2.44(m,1h),1.91(m,2h),1.53(m,2h).ms(esi):m/z 441.0[m h]
。
[0139]
实施例7化合物7的制备
[0140][0141]
第一步:化合物7b的制备
[0142]
室温下将6-甲基吡啶-3-醇7a(0.57g,5.2mmol),3,4,5-三氟苯甲醛5a(1g,6.2mmol)和碳酸钾(0.93g,6.76mmol)溶于30ml n,n-二甲基甲酰胺中,在90℃搅拌反应1小时,冷却到室温后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=10/1)纯化得到黄色固体产物7b(0.91g,产率:69.2%)。
[0143]1h nmr(400mhz,cdcl3)δ9.92(s,1h),8.28(s,1h),7.62
–
7.49(m,2h),7.18
–
7.10(m,2h),2.54(s,3h)。
[0144]
第二步:化合物7c的制备
[0145]
室温下将3,5-二氟-4-((6-甲基吡啶-3-基)氧基)苯甲醛7b(0.91g,3.6mmol)溶于50ml甲醇中,在0℃加入nabh4(161mg,4.3mmol),室温搅拌反应0.5小时,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=4/1)纯化得到无色油状产物7c(0.89g,产率:98.4%)。
[0146]1h nmr(400mhz,cdcl3)δ8.20(m,1h),7.16
–
6.98(m,4h),4.69(m,2h),2.88(m,1h),2.50(s,3h)。
[0147]
第三步:化合物7的制备
[0148]
将(3,5-二氟-4
–
((6-甲基吡啶-3-基)氧基)苯基)甲醇7c(56mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物7(17mg,17.5%)。
[0149]1h nmr(400mhz,cdcl3)δ8.27(m,1h),7.10(m,4h),5.62(s,1h),5.39(s,2h),4.07(m,7.3hz,1h),3.80(m,1h),3.46(m,1h),3.29
–
3.21(m,1h),3.15(m,1h),3.04(m,1h),2.50(s,3h),2.43(m,1h),1.97
–
1.86(m,2h),1.51(m,2h).ms(esi):m/z441.0[m h]
。
[0150]
实施例8化合物8的制备
[0151][0152]
第一步:化合物8b的制备
[0153]
室温下将2-甲基嘧啶-5-醇8a(0.25g,2.3mmol),3,4,5-三氟苯甲醛5a(0.44g,2.8mmol)和碳酸钾(0.41g,2.9mmol)溶于30ml n,n-二甲基甲酰胺中,在90℃搅拌反应2小时,冷却下来后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=10/1)纯化得到黄色固体产物8b(0.24g,产率:41.7%)。
[0154]1h nmr(400mhz,cdcl3)δ9.93(s,1h),8.39(s,2h),7.64
–
7.54(m,2h),2.72(s,3h)。
[0155]
第二步:化合物8c的制备
[0156]
室温下将3,5-二氟-4-((2-甲基嘧啶-5-基)氧基)苯甲醛8b(0.24g,0.96mmol)溶于50ml甲醇中,在0℃加入nabh4(30mg,0.79mmol),室温搅拌反应0.5小时,减压浓缩,加入
水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用氯化钠溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=4/1)纯化得到无色油状产物8c(0.17g,产率:70.2%)。
[0157]1h nmr(400mhz,cdcl3)δ8.33(s,2h),7.04(m,2h),4.71(m,2h),2.70(s,3h)。
[0158]
第三步:化合物8的制备
[0159]
将(3,5-二氟-4
–
((2-甲基嘧啶-5-基)氧基)苯基)甲醇8c(56mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物8(10mg,10.3%)。
[0160]1h nmr(400mhz,cdcl3)δ8.35(s,2h),7.11(m,2h),5.62(s,1h),5.40(s,2h),4.06(s,1h),3.80(m,1h),3.47(m,1h),3.25(s,1h),3.15(m,1h),3.04(m,1h),2.71(s,3h),2.44(m,1h),1.91(m,2h),1.52(m,2h).ms(esi):m/z 442.0[m h]
。
[0161]
实施例9化合物9的制备
[0162][0163][0164]
第一步:化合物9b的制备
[0165]
室温下将2-(三氟甲基)嘧啶-5-醇9a(0.25g,1.52mmol),3,4,5-三氟苯甲醛5a(0.29g,1.81mmol)和碳酸钾(0.27g,1.98mmol)溶于20ml n,n-二甲基甲酰胺中,在90℃搅拌反应2小时,冷却下来后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=10/1)纯化得到黄色固体产物9b(0.24g,产率:51.9%)。
[0166]1h nmr(400mhz,cdcl3)δ9.97(s,1h),8.59(s,2h),7.69
–
7.54(m,2h)。
[0167]
第二步:化合物9c的制备
[0168]
室温下将3,5-二氟-4-((2-(三氟甲基)嘧啶-5-基)氧基)苯甲醛9b(0.24g,0.79mmol)溶于50ml甲醇中,在0℃加入nabh4(30mg,0.79mmol),室温搅拌反应0.5h,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=4/1)纯化得到无色油状产物9c(0.12g,产率:49.6%)。
[0169]1h nmr(400mhz,cdcl3)δ8.54(s,2h),7.12(m,2h),4.74(m,2h),2.23(m,1h)。
[0170]
第三步:化合物9的制备
[0171]
将(3,5-二氟-4-((2-(三氟甲基)嘧啶-5-基)氧基)苯基)甲醇9c(73mg,0.24mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物9(8mg,7.3%)。
[0172]1h nmr(400mhz,cdcl3)δ8.58(s,2h),7.19(m,2h),5.66(s,1h),5.45(s,2h),4.10(m,7.4hz,1h),3.82(m,1h),3.49(m,1h),3.34
–
3.23(m,1h),3.18(m,1h),3.07(m,1h),2.47(m,1h),1.97(m,2h),1.60
–
1.50(m,2h).ms(esi):m/z 496.1[m h]
。
[0173]
实施例10化合物10的制备
[0174][0175]
第一步:化合物10b的制备
[0176]
室温下将3-(三氟甲基)苯酚10a(1g,6.2mmol),3,4,5-三氟苯甲醛4a(1.09g,6.8mmol)和碳酸钾(1.1g,8.02mmol)溶于30ml n,n-二甲基甲酰胺中,在90℃搅拌反应2小时,冷却下来后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=10/1)纯化得到黄色固体产物10b(1.7g,产率:90.7%)。
[0177]1h nmr(400mhz,cdcl3)δ9.94(s,1h),7.63
–
7.55(m,2h),7.46(m,1h),7.39(m,1h),7.21(s,1h),7.13(m,1h)。
[0178]
第二步:化合物10c的制备
[0179]
室温下将3,5-二氟-4-(3-(三氟甲基)苯氧基)苯甲醛10b(1.7g,5.6mmol)溶于50ml甲醇中,在0℃加入nabh4(213mg,5.6mmol),室温搅拌反应0.5小时,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=4/1)纯化得到无色油状产物10c(1.27g,产率:74.5%)。
[0180]
第三步:化合物10的制备
[0181]
将(3,5-二氟-4-(3-(三氟甲基)苯氧基)苯基)甲醇10c(73mg,0.24mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭
反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物10(6mg,5.5%)。
[0182]1h nmr(400mhz,cdcl3)δ7.42(m,1h),7.33(m,1h),7.20(s,1h),7.10(m,3h),5.63(s,1h),5.41(s,2h),4.11
–
4.02(m,1h),3.80(m,1h),3.47(m,1h),3.26(m,1h),3.15(m,1h),3.04(m,1h),2.43(m,1h),2.00
–
1.85(m,2h),1.55
–
1.48(m,2h).ms(esi):m/z494.0[m h]
。
[0183]
实施例11化合物11的制备
[0184][0185]
第一步:化合物11b的制备
[0186]
室温下将4-氯-3-甲基苯酚11a(1g,7.0mmol),3,4,5-三氟苯甲醛5a(1.2g,7.5mmol)和碳酸钾(1.3g,9.1mmol)溶于30ml n,n-二甲基甲酰胺中,在90℃搅拌反应2小时,冷却下来后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=10/1)纯化得到黄色固体产物11b(1.2g,产率:60.6%)。
[0187]1h nmr(400mhz,cdcl3)δ9.92(s,1h),7.61
–
7.51(m,2h),7.30
–
7.23(m,1h),6.85(m,1h),6.73(m,1h),2.34(s,3h)。
[0188]
第二步:化合物11c的制备
[0189]
室温下将4-(4-氯-3-甲基苯氧基)-3,5-二氟苯甲醛11b(1.2g,4.2mmol)溶于50ml甲醇中,在0℃加入nabh4(161mg,4.2mmol),室温搅拌反应0.5小时,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=4/1)纯化得到无色油状产物11c(0.89g,产率:74.4%)。
[0190]
第三步:化合物11的制备
[0191]
将4-(4-氯-3-甲基苯氧基)-3,5-二氟苯甲醇11c(77mg,0.27mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.42mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物11(9mg,8.6%)。
[0192]1h nmr(400mhz,cdcl3)δ7.27(m,1h),7.12(m,2h),6.86(m,1h),6.74(m,1h),5.66
(s,1h),5.43(s,2h),4.11(m,1h),3.84(m,1h),3.50(m,1h),3.34
–
3.25(m,1h),3.19(m,1h),3.08(m,1h),2.47(m,1h),2.36(s,3h),1.98(m,2h),1.61
–
1.51(m,2h).
[0193]
实施例12化合物12的制备
[0194][0195]
第一步:化合物12b的制备
[0196]
室温下将3-(三氟甲氧基)苯酚12a(0.50g,2.8mmol),3,4,5-三氟苯甲醛5a(0.5g,3.1mmol)和碳酸钾(0.5g,3.64mmol)溶于30ml n,n-二甲基甲酰胺中,在90℃搅拌反应2小时,冷却下来后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=10/1)纯化得到黄色固体产物12b(0.73g,产率:91.8%)。
[0197]1h nmr(400mhz,cdcl3)δ9.94(s,1h),7.64
–
7.54(m,2h),7.34(m,1h),7.00(m,1h),6.87(m,2h)。
[0198]
第二步:化合物12c的制备
[0199]
室温下将4-(3-(三氟甲氧基)苯氧基)-3,5-二氟苯甲醛12b(0.73g,2.3mmol)溶于50ml甲醇中,在0℃加入nabh4(86mg,2.28mmol),室温搅拌反应0.5小时,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=4/1)纯化得到无色油状产物12c(0.57g,产率:77.4%)。
[0200]1h nmr(400mhz,cdcl3)δ7.30(m,1h),7.06(m,2h),6.94(m,1h),6.85(m,1h),6.81(s,1h),4.72(m,2h),1.94(m,1h)。
[0201]
第三步:化合物12的制备
[0202]
将4-(3-(三氟甲氧基)苯氧基)-3,5-二氟苯基)甲醇12c(70mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物12(6mg,5.4%)。
[0203]1h nmr(400mhz,cdcl3)δ7.29(m,1h),7.08(m,2h),6.93(m,1h),6.88
–
6.77(m,2h),5.62(s,1h),5.39(s,2h),4.06(m,1h),3.81(m,1h),3.46(m,1h),3.31
–
3.20(m,1h),3.13(m,1h),3.04(m,1h),2.43(m,1h),1.89(m,2h),1.51(m,2h).ms(esi):m/z 510.0[m h]
。
[0204]
实施例13化合物13的制备
[0205][0206]
第一步:化合物13b的制备
[0207]
室温下将4-(三氟甲基)苯酚13a(0.84g,5.2mmol),3,4,5-三氟苯甲醛5a(1g,6.2mmol)和碳酸钾(0.93g,6.76mmol)溶于30ml n,n-二甲基甲酰胺中,在90℃搅拌反应1小时,冷却到室温倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=5/1)纯化得到黄色固体产物13b(1.33g,产率:84.6%)。
[0208]1h nmr(400mhz,cdcl3)δ9.94(m,1h),7.59(m,4h),7.04(m,2h)。
[0209]
第二步:化合物13c的制备
[0210]
室温下将3,5-二氟-4-(4-(三氟甲基)苯氧基)苯甲醛13b(1.33g,4.4mmol)溶于50ml甲乙醇中,在0℃加入nabh4(166mg,4.4mmol),室温搅拌反应0.5小时,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=2/1)纯化得到无色油状产物13c(0.85g,产率:63.5%)。
[0211]1h nmr(400mhz,cdcl3)δ7.57(m,2h),7.09
–
7.00(m,4h),4.72(m,2h),2.03(m,1h)。
[0212]
第三步:化合物13的制备
[0213]
将3,5-二氟-4-(4-(三氟甲基)苯氧基)苯基)甲醇13c(67mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物13(11mg,10.1%)。
[0214]1h nmr(400mhz,cdcl3)δ7.57(m,2h),7.10(m,2h),7.01(m,2h),5.63(s,1h),5.41(s,2h),4.11
–
4.04(m,1h),3.80(m,1h),3.47(m,1h),3.30
–
3.21(m,1h),3.16(m,1h),3.04(m,1h),2.44(m,1h),1.99
–
1.85(m,2h),1.57
–
1.48(m,2h).ms(esi):m/z494.0[m h]
。
[0215]
实施例14化合物14的制备
[0216][0217]
第一步:化合物14b的制备
[0218]
室温下将4-氯-3-(三氟甲基)苯酚14a(0.5g,2.5mmol),3,4,5-三氟苯甲醛5a(0.45g,2.8mmol)和碳酸钾(0.46g,3.3mmol)溶于30ml n,n-二甲基甲酰胺中,在90℃搅拌反应2小时,冷却下来后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=10/1)纯化得到黄色固体产物14b(0.6g,产率:71.3%)。
[0219]1h nmr(400mhz,cdcl3)δ9.94(s,1h),7.64
–
7.55(m,2h),7.45(m,1h),7.31(m,1h),7.05(m,1h)。
[0220]
第二步:化合物14c的制备
[0221]
室温下将4-(4-氯-3-(三氟甲基)苯氧基)-3,5-二氟苯甲醛14b(0.6g,1.78mmol)溶于50ml甲醇中,在0℃加入nabh4(67mg,1.78mmol),室温搅拌反应0.5小时,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=4/1)纯化得到白色固体状产物14c(0.28g,产率:46.4%)。
[0222]1h nmr(400mhz,cdcl3)δ7.41(m,1h),7.28(m,1h),7.08
–
7.00(m,3h),4.73(m,2h),1.94(m,1h)。
[0223]
第三步:化合物14的制备
[0224]
将(4-(4-氯-3-(三氟甲基)苯氧基)-3,5-二氟苯基)甲醇14c(74 mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物14(22mg,18.9%)。
[0225]1h nmr(400mhz,cdcl3)δ7.41(m,1h),7.30(m,1h),7.11(m,2h),7.01(m,1h),5.63(s,1h),5.41(s,2h),4.07(m,1h),3.80(m,1h),3.47(m,1h),3.30
–
3.21(m,1h),3.15(m,1h),3.04(m,1h),2.44(m,1h),1.95(m,2h),1.57
–
1.47(m,2h).
[0226]
实施例15化合物15的制备
[0227][0228][0229]
第一步:化合物15b的制备
[0230]
室温下将3-氯-4-(三氟甲氧基)苯酚15a(0.50g,2.4mmol),3,4,5-三氟苯甲醛5a(0.41g,2.6mmol)和碳酸钾(0.42g,3.04mmol)溶于20ml n,n-二甲基甲酰胺中,在90℃搅拌反应2小时,冷却下来后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=10/1)纯化得到黄色固体产物15b(0.62g,产率:73.2%)。
[0231]1h nmr(400mhz,cdcl3)δ9.94(s,1h),7.63
–
7.54(m,2h),7.29(m,1h),7.07(m,1h),6.90(m,1h)。
[0232]
第二步:化合物15c的制备
[0233]
室温下将4-(3-氯-4-(三氟甲氧基)苯氧基)-3,5-二氟苯甲醛15b(0.62g,1.8mmol)溶于50ml甲醇中,在0℃加入nabh4(62mg,1.94mmol),室温搅拌反应0.5小时,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=4/1)纯化得到无色油状产物15c(0.53g,产率:83.0%)。
[0234]1h nmr(400mhz,cdcl3)δ7.25(m,1h),7.06(m,2h),7.01(m,1h),6.87(m,1h),4.72(s,2h),2.04(m,1h)。
[0235]
第三步:化合物15的制备
[0236]
将(4-(3-氯-4-(三氟甲氧基)苯氧基)-3,5-二氟苯基)甲醇15c(79mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物15(7mg,5.8%)。
[0237]1h nmr(400mhz,cdcl3)δ7.24(m,1h),7.10(m,2h),7.03(m,1h),6.87(m,1h),5.63(s,1h),5.41(s,2h),4.10
–
4.03(m,1h),3.80(m,1h),3.47(m,1h),3.30
–
3.21(m,1h),3.16(m,1h),3.04(m,1h),2.44(m,1h),1.95(m,2h),1.56
–
1.49(m,2h).
[0238]
实施例16化合物16的制备
[0239][0240]
第一步:化合物16b的制备
[0241]
室温下将3-氯-4-(三氟甲基)苯酚16a(0.25g,1.23mmol),3,4,5-三氟苯甲醛5a(0.22g,1.4mmol)和碳酸钾(0.23g,1.65mmol)溶于20ml n,n-二甲基甲酰胺中,在90℃搅拌反应2小时,冷却下来后倒入100ml冰水,用乙酸乙酯萃取(50ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=10/1)纯化得到黄色固体产物16b(0.32g,产率:77.3%)。
[0242]1h nmr(400mhz,cdcl3)δ9.95(s,1h),7.69
–
7.56(m,3h),7.10(m,1h),6.92(m,1h)。
[0243]
第二步:化合物16c的制备
[0244]
室温下将4-(3-氯-4-(三氟甲基)苯氧基)-3,5-二氟苯甲醛16b(0.32g,0.95mmol)溶于50ml甲醇中,在0℃加入nabh4(36mg,0.94mmol),室温搅拌反应0.5小时,减压浓缩,加入水,用乙酸乙酯萃取(100ml
×
2),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(石油醚/乙酸乙酯=4/1)纯化得到白色固体状产物16c(0.15g,产率:46.6%)。
[0245]1h nmr(400mhz,cdcl3)δ7.62(m,1h),7.13
–
7.00(m,3h),6.90(m,1h),4.74(m,2h),1.88(m,1h)。
[0246]
第三步:化合物16的制备
[0247]
将(4-(3-氯-4-(三氟甲基)苯氧基)-3,5-二氟苯基)甲醇(74.5mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.44mmol),室温下搅拌反应5分钟后,加入化合物1f(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物16(13mg,11.2%)。
[0248]1h nmr(400mhz,cdcl3)δ7.62(m,1h),7.12(m,2h),7.07(m,1h),6.90(m,1h),5.63(s,1h),5.42(s,2h),4.12
–
4.03(m,1h),3.81(m,1h),3.45(m,1h),3.32
–
3.21(m,1h),3.16(m,1h),3.05(m,1h),2.44(m,1h),1.91(m,2h),1.56
–
1.49(m,2h).
[0249]
实施例17化合物17的制备
[0250][0251]
第一步:化合物17b的制备
[0252][0253]
室温下将6-氯尿嘧啶(8.2g,55.9mmol)、(s)-3-(羟甲基)哌啶-1-甲酸叔丁酯17a(12g,55.7mmol)和三苯基膦(20g,76.2mmol)溶于250ml的无水四氢呋喃和50ml的无水n,n-二甲基甲酰胺混合溶剂中,氮气保护和0℃下滴加偶氮二甲酸二异丙酯(20ml,111.6mmol),在0℃下搅拌反应2小时后,升温到室温反应过夜,过滤反应液,用乙酸乙酯萃取(50ml
×
3),合并有机相,饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到无色油状产物17b(7.8g,产率:40.6%)。
[0254]
第二、三步:化合物17d的制备
[0255][0256]
室温下将化合物(s)-3-(((6-氯-2,4-二氧代-3,4-二氢嘧啶-1(2h)-基)甲基)哌啶-1-甲酸叔丁酯17b(7.8g,22.7mmol)溶于80ml的二氯甲烷中,0℃下加入10ml的三氟乙酸,室温下搅拌反应2小时,将反应液减压浓缩,直接进行下一步反应。将上一步的粗品溶于80ml乙腈中,室温下加入二异丙基乙胺(8.8g,68.1mmol),搅拌反应4小时,减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物17d(1.5g,31.9%)。
[0257]1h nmr(400mhz,cdcl3)δ9.51(s,1h),5.35(s,1h),4.03(m,1h),3.78
–
3.64(m,1h),3.43(m,1h),3.24(m,1h),3.18
–
2.96(m,2h),2.37(m,1h),2.00
–
1.83(m,2h),1.72
–
1.49(m,2h).
[0258]
第四步:化合物17e的制备
[0259]
[0260]
室温下将化合物17d(0.51g,2.46mmol)和二甲基苯胺(0.3g,2.47mmol)溶于甲苯中,滴加三氯氧磷(0.76g,4.96mmol),加热回流4小时后,用冰水淬灭反应,减压浓缩,用乙酸乙酯萃取(60ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物17e(0.24g,43.2%)。
[0261]1h nmr(400mhz,cdcl3)δ6.05(s,1h),4.04(m,1h),3.74(m,1h),3.51(m,1h),3.28(m,1h),3.20
–
3.14(m,1h),3.01(m,1h),2.51(m,1h),1.92(m,2h),1.59
–
1.41(m,2h).
[0262]
第五步:化合物17的制备
[0263][0264]
将(3,5-二氟-4
–
((2-(三氟甲基吡啶-4-基)氧基)苯基)甲醇5d(67mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,18mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物17e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物17(28mg,25.7%)。
[0265]1h nmr(400mhz,cdcl3)δ8.59(m,1h),7.25(s,1h),7.15(m,2h),6.98(m,1h),5.63(s,1h),5.42(s,2h),4.07(m,1h),3.80(m,1h),3.46(m,1h),3.27(m,1h),3.16(m,1h),3.03(m,1h),2.44(m,1h),1.90(m,2h),1.52(m,2h).ms(esi):m/z 495.1[m h]
。
[0266]
实施例18化合物18的制备
[0267][0268]
将(3,5-二氟-4
–
((6-甲基吡啶-3-基)氧基)苯基)甲醇7c(56mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物17e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物18(22mg,22.7%)。
[0269]1h nmr(400mhz,cdcl3)δ8.27(m,1h),7.10(m,4h),5.62(s,1h),5.39(s,2h),4.07(m,1h),3.80(m,1h),3.46(m,1h),3.29
–
3.21(m,1h),3.15(m,1h),3.04(m,1h),2.51(s,3h),2.44(m,1h),1.99
–
1.84(m,2h),1.51(m,2h).ms(esi):m/z 441.2[m h]
。
[0270]
实施例19化合物19的制备
[0271]
[0272]
将(3,5-二氟-4
–
((2-甲基)吡啶-4-基)氧基)苯基)甲醇6c(56mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物17e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物19(58mg,59.8%)。
[0273]1h nmr(400mhz,cdcl3)δ8.35(m,1h),7.10(m,2h),6.67(m,2h),5.62(s,1h),5.41(s,2h),4.07(m,1h),3.79(m,1h),3.46(m,1h),3.30
–
3.18(m,1h),3.15(m,1h),3.04(m,1h),2.50(s,3h),2.43(m,1h),1.91(m,2h),1.55
–
1.46(m,2h).ms(esi):m/z 441.2[m h]
。
[0274]
实施例20化合物20的制备
[0275][0276]
将(3,5-二氟-4-(3-(三氟甲基)苯氧基)苯基)甲醇10c(67mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物17e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物20(46mg,42.4%)。
[0277]1h nmr(400mhz,cdcl3)δ7.41(m,1h),7.32(m,1h),7.19(s,1h),7.09(m,3h),5.62(s,1h),5.40(s,2h),4.06(m,1h),3.79(m,1h),3.46(m,1h),3.30
–
3.19(m,1h),3.14(m,1h),3.03(m,1h),2.43(m,1h),1.98
–
1.84(m,2h),1.57
–
1.46(m,2h).ms(esi):m/z494.1[m h]
。
[0278]
实施例21化合物21的制备
[0279][0280]
将3,5-二氟苯甲醇(32mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物17e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物21(17mg,23.2%)。
[0281]1h nmr(400mhz,cdcl3)δ6.96
–
6.87(m,2h),6.74(m,1h),5.60(s,1h),5.37(s,2h),4.06(m,1h),3.79(m,1h),3.45(m,1h),3.23(m,1h),3.14(m,1h),3.03(m,1h),2.42(m,1h),1.99
–
1.83(m,2h),1.57
–
1.46(m,2h).ms(esi):m/z 334.1[m h]
。
[0282]
实施例22化合物22的制备
[0283][0284]
将2,4,5-三氟苯甲醇(36mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物17e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物22(36mg,46.6%)。
[0285]1h nmr(400mhz,cdcl3)δ7.32(m,1h),6.94(m,1h),5.57(s,1h),5.40(s,2h),4.06(m,1h),3.79(m,1h),3.44(m,1h),3.29
–
3.17(m,1h),3.14(m,1h),3.03(m,1h),2.42(m,1h),1.99
–
1.83(m,2h),1.55
–
1.45(m,2h).ms(esi):m/z 352.1[m h]
。
[0286]
实施例23化合物23的制备
[0287][0288]
第一步:化合物23b的制备
[0289][0290]
室温下将6-氯尿嘧啶(6.1g,41.6mmol)、(r)-3-(羟甲基)哌啶-1-甲酸叔丁酯23a(9g,41.8mmol)和三苯基膦(16.3g,62.1mmol)溶于250ml的无水四氢呋喃和50ml的无水n,n-二甲基甲酰胺混合溶剂中,氮气保护和0℃下滴加偶氮二甲酸二异丙酯(16ml,83mmol),在0℃下搅拌反应2小时后,升温到室温反应过夜,过滤反应液,用乙酸乙酯萃取(50ml
×
3),合并有机相,饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到无色油状产物23b(8.3g,产率:58.0%)。
[0291]
第二、三步:化合物23d的制备
[0292][0293]
室温下将化合物(r)-3-(((6-氯-2,4-二氧代-3,4-二氢嘧啶-1(2h)-基)甲基)哌啶-1-甲酸叔丁酯23b(8.3g,24.1mmol)溶于80ml的二氯甲烷中,0℃下加入10ml的三氟乙
酸,室温下搅拌反应2小时,将反应液减压浓缩,直接进行下一步反应。将上一步的粗品溶于80ml乙腈中,室温下加入碳酸钾(7.0g,50.9mmol),加热回流反应过夜,过滤,滤饼分别用乙腈和甲醇冲洗,将滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物23d(1.5g,29.9%)。
[0294]1h nmr(400mhz,cdcl3)δ8.85(s,1h),5.33(s,1h),4.01(m,1h),3.70(m,1h),3.42(m,1h),3.23(m,1h),3.11(m,1h),3.03(m,1h),2.35(m,1h),1.99
–
1.82(m,2h),1.67
–
1.47(m,2h).
[0295]
第四步:化合物23e的制备
[0296][0297]
室温下将化合物23d(0.5g,2.4mmol)和二甲基苯胺(0.29g,2.4mmol)溶于甲苯中,滴加三氯氧磷(0.74g,4.8mmol),加热回流4小时后,用冰水淬灭反应,减压浓缩,用乙酸乙酯萃取(60ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物23e(0.31g,57.2%)。
[0298]1h nmr(400mhz,cdcl3)δ6.05(s,1h),4.05(m,1h),3.76(m,1h),3.51(m,1h),3.28(m,1h),3.22
–
3.13(m,1h),3.02(m,1h),2.52(m,1h),2.01
–
1.82(m,2h),1.61
–
1.39(m,2h).
[0299]
第五步:化合物23的制备
[0300][0301]
将(3,5-二氟-4
–
((2-(三氟甲基吡啶-4-基)氧基)苯基)甲醇5d(67mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物23e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物23(25mg,23.0%)。
[0302]1h nmr(400mhz,cdcl3)δ8.59(m,1h),7.25(s,1h),7.14(m,2h),6.98(m,1h),5.63(s,1h),5.42(s,2h),4.07(m,1h),3.79(m,1h),3.46(m,1h),3.26(m,1h),3.15(m,1h),3.04(m,1h),2.44(m,1h),2.00
–
1.84(m,2h),1.56
–
1.46(m,2h).ms(esi):m/z 495.1[m h]
。
[0303]
实施例24化合物24的制备
[0304][0305]
将(3,5-二氟-4
–
((6-甲基吡啶-3-基)氧基)苯基)甲醇7c(56mg,0.22mmol)溶于
5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物23e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物24(52mg,53.7%)。
[0306]1h nmr(400mhz,cdcl3)δ8.27(m,1h),7.09(m,4h),5.61(s,1h),5.38(s,2h),4.06(m,1h),3.80(m,1h),3.46(m,1h),3.28
–
3.20(m,1h),3.15(m,1h),3.04(m,1h),2.51(s,3h),2.43(m,1h),1.92(m,2h),1.57
–
1.45(m,2h).ms(esi):m/z 441.2[m h]
。
[0307]
实施例25化合物25的制备
[0308][0309]
将(3,5-二氟-4
–
((2-甲基)吡啶-4-基)氧基)苯基)甲醇6c(56mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物23e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物25(38mg,39.2%)。
[0310]1h nmr(400mhz,cdcl3)δ8.36(m,1h),7.11(m,2h),6.67(m,2h),5.63(s,1h),5.41(s,2h),4.07(m,1h),3.80(m,1h),3.47(m,1h),3.32
–
3.21(m,1h),3.15(m,1h),3.04(m,1h),2.51(s,3h),2.44(m,1h),2.00
–
1.83(m,2h),1.59
–
1.46(m,2h).ms(esi):m/z441.2[m h]
。
[0311]
实施例26化合物26的制备
[0312][0313]
将(3,5-二氟-4-(3-(三氟甲基)苯氧基)苯基)甲醇10c(67mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物23e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物26(58mg,53.4%)。
[0314]1h nmr(400mhz,cdcl3)δ7.42(m,1h),7.33(m,1h),7.20(s,1h),7.11(m,3h),5.63(s,1h),5.41(s,2h),4.07(m,1h),3.80(m,1h),3.47(m,1h),3.30
–
3.20(m,1h),3.15(m,1h),3.04(m,1h),2.43(m,1h),1.98
–
1.84(m,2h),1.56
–
1.48(m,2h).ms(esi):m/z494.1[m h]
。
[0315]
实施例27化合物27的制备
二甲基甲酰胺混合溶剂中,氮气保护和0℃下滴加偶氮二甲酸二异丙酯(27ml,138mmol),在0℃下搅拌反应2小时后,升温到室温反应过夜,过滤反应液,用乙酸乙酯萃取(50ml
×
3),合并有机相,饱和氯化钠水溶液洗涤,无水硫酸钠干燥,减压浓缩,加入100ml pe/ea=3/1,有大量白色固体析出,过滤,将反应液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到无色油状产物29b(11.5g,产率:48.2%)。
[0328]
第二、三步:化合物29d的制备
[0329][0330]
室温下将化合物(s)-2-(((6-氯-2,4-二氧代-3,4-二氢嘧啶-1(2h)-基)甲基)吗啉-4-羧酸叔丁酯29b(11.5g,33.2mmol)溶于80ml的二氯甲烷中,0℃下加入10ml的三氟乙酸,室温下搅拌反应2小时,将反应液减压浓缩,直接进行下一步反应。将上一步的粗品溶于80ml乙腈中,室温下加入碳酸钾(9.2g,67mmol),加热回流反应过夜,过滤,滤饼分别用乙腈和甲醇冲洗,将滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物29d(1.9g,27.4%)。
[0331]1h nmr(400mhz,cdcl3)δ9.39(s,1h),5.41(s,1h),4.36
–
4.28(m,1h),4.05(m,1h),3.95(m,1h),3.78(m,1h),3.69
–
3.55(m,2h),3.44
–
3.37(m,1h),3.24(m,1h),2.96(m,1h).
[0332]
第四步:化合物29e的制备
[0333][0334]
室温下将化合物29d(0.5g,2.4mmol)和二甲基苯胺(0.29g,2.4mmol)溶于甲苯中,滴加三氯氧磷(0.73g,4.8mmol),加热回流4小时后,用冰水淬灭反应,减压浓缩,用乙酸乙酯萃取(60ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物29e(0.3g,54.9%)。
[0335]1h nmr(400mhz,cdcl3)δ6.12(s,1h),4.49(m,1h),4.08(m,1h),3.99(m,1h),3.72
–
3.61(m,3h),3.48(m,1h),3.37(m,1h),2.96(m,1h).
[0336]
第五步:化合物29的制备
[0337][0338]
将(3,5-二氟-4
–
((2-(三氟甲基吡啶-4-基)氧基)苯基)甲醇5d(67mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物29e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白
色固体产物29(31mg,28.4%)。
[0339]1h nmr(400mhz,cdcl3)δ8.59(m,1h),7.25(s,1h),7.14(m,2h),6.98(m,1h),5.69(s,1h),5.43(s,2h),4.40(m,1h),4.12(m,1h),4.03(m,1h),3.70
–
3.57(m,3h),3.44(m,1h),3.27(m,1h),2.95(m,1h).ms(esi):m/z 497.1[m h]
。
[0340]
实施例30化合物30的制备
[0341][0342]
将(3,5-二氟-4
–
((6-甲基吡啶-3-基)氧基)苯基)甲醇(56mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物29e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物30(49mg,50.3%)。
[0343]1h nmr(400mhz,cdcl3)δ8.26(m,1h),7.16
–
7.05(m,4h),5.69(s,1h),5.40(s,2h),4.40(m,1h),4.13(m,1h),4.04(m,1h),3.70
–
3.58(m,3h),3.45(m,1h),3.27(m,1h),2.96(m,1h),2.52(s,3h).ms(esi):m/z443.1[m h]
。
[0344]
实施例31化合物31的制备
[0345][0346]
将(3,5-二氟-4
–
((2-甲基)吡啶-4-基)氧基)苯基)甲醇6c(56mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物29e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物31(38mg,39.0%)。
[0347]1h nmr(400mhz,cdcl3)δ8.35(m,1h),7.11(m,2h),6.72
–
6.63(m,2h),5.70(s,1h),5.42(s,2h),4.39(m,1h),4.17
–
4.08(m,1h),4.03(m,1h),3.71
–
3.57(m,3h),3.50
–
3.40(m,1h),3.27(m,1h),2.95(m,1h),2.50(s,3h).ms(esi):m/z 443.1[m h]
。
[0348]
实施例32化合物32的制备
[0349][0350]
将(3,5-二氟-4-(3-(三氟甲基)苯氧基)苯基)甲醇10c(67mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物29e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭
反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物32(31mg,28.4%)。
[0351]1h nmr(400mhz,cdcl3)δ7.42(m,1h),7.34(m,1h),7.19(s,1h),7.10(m,3h),5.70(s,1h),5.42(s,2h),4.40(m,1h),4.17
–
4.00(m,2h),3.71
–
3.57(m,3h),3.45(m,1h),3.28(m,1h),2.96(m,1h).ms(esi):m/z 496.1[m h]
。
[0352]
实施例33化合物33的制备
[0353][0354]
将3,5-二氟苯甲醇(32mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物29e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物33(21mg,28.5%)。
[0355]1h nmr(400mhz,cdcl3)δ6.98
–
6.91(m,2h),6.77(m,1h),5.70(s,1h),5.40(s,2h),4.41(m,1h),4.14(m,1h),4.04(m,1h),3.72
–
3.58(m,3h),3.46(m,1h),3.28(m,1h),2.97(m,1h).ms(esi):m/z 336.1[m h]
。
[0356]
实施例34化合物34的制备
[0357][0358]
将2,4,5-三氟苯甲醇(36mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物29e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物34(29mg,37.3%)。
[0359]1h nmr(400mhz,cdcl3)δ7.36
–
7.27(m,1h),6.95(m,1h),5.64(s,1h),5.40(s,2h),4.42
–
4.35(m,1h),4.12(m,1h),4.02(m,1h),3.73
–
3.55(m,3h),3.43(m,1h),3.24(m,1h),2.95(m,1h).ms(esi):m/z 354.1[m h]
。
[0360]
实施例35化合物35的制备
[0361][0362]
第一步:化合物35b的制备
[0363][0364]
室温下将6-氯尿嘧啶(10g,68.2mmol)、(r)-2-(羟甲基)吗啉-4-羧酸叔丁酯35a(15g,69mmol)和三苯基膦(27g,102.9mmol)溶于250ml的无水四氢呋喃和50ml的无水n,n-二甲基甲酰胺混合溶剂中,氮气保护和0℃下滴加偶氮二甲酸二异丙酯(27ml,138mmol),在0℃下搅拌反应2小时后,升温到室温反应过夜,过滤反应液,用乙酸乙酯萃取(50ml
×
3),合并有机相,饱和氯化钠水溶液洗涤,无水硫酸钠干燥,减压浓缩,加入100ml pe/ea=3/1,有大量白色固体析出,过滤,将滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到无色油状产物35b(7.2g,产率:30.5%)。
[0365]
第二、三步:化合物35d的制备
[0366][0367]
室温下将化合物(r)-2-(((6-氯-2,4-二氧代-3,4-二氢嘧啶-1(2h)-基)甲基)吗啉-4-羧酸叔丁酯35b(7.2g,20.8mmol)溶于80ml的二氯甲烷中,0℃下加入10ml的三氟乙酸,室温下搅拌反应2小时,将反应液减压浓缩,直接进行下一步反应。将上一步的粗品溶于80ml乙腈中,室温下加入二异丙基乙胺(8.1g,62.7mmol),室温下搅拌反应4小时,将反应液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物35d(2.5g,57.4%)。
[0368]1h nmr(400mhz,cdcl3)δ9.32(s,1h),5.42(s,1h),4.35
–
4.29(m,1h),4.07(m,1h),3.96(m,1h),3.78(m,1h),3.70
–
3.55(m,2h),3.45
–
3.36(m,1h),3.24(m,1h),2.96(m,1h).
[0369]
第四步:化合物35e的制备
[0370][0371]
室温下将化合物35d(0.1g,0.48mmol)和二甲基苯胺(0.058g,0.48mmol)溶于甲苯中,滴加三氯氧磷(0.73g,4.8mmol),加热回流4小时后,用冰水淬灭反应,减压浓缩,用乙酸乙酯萃取(60ml
×
3),合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤除去干燥剂,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物35e(0.04g,36.4%)。
[0372]1h nmr(400mhz,cdcl3)δ6.12(s,1h),4.48(m,1h),4.06(m,1h),3.99(m,1h),3.73
–
3.61(m,3h),3.47(m,1h),3.42
–
3.32(m,1h),2.96(m,1h).
[0373]
第五步:化合物35的制备
[0374][0375]
将(3,5-二氟-4
–
((2-(三氟甲基吡啶-4-基)氧基)苯基)甲醇5d(67mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物35e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物35(41mg,37.5%)。
[0376]1h nmr(400mhz,cdcl3)δ8.60(m,1h),7.25(s,1h),7.15(m,2h),6.99(m,1h),5.70(s,1h),5.44(s,2h),4.40(m,1h),4.13(m,1h),4.04(m,1h),3.70
–
3.58(m,3h),3.44(m,1h),3.28(m,1h),2.96(m,1h).ms(esi):m/z 497.1[m h]
。
[0377]
实施例36化合物36的制备
[0378][0379]
将(3,5-二氟-4
–
((6-甲基吡啶-3-基)氧基)苯基)甲醇7c(56mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物35e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物36(42mg,43.2%)。
[0380]1h nmr(400mhz,cdcl3)δ8.21(m,1h),7.06(m,4h),5.65(s,1h),5.37(s,2h),4.36(m,1h),4.13
–
4.03(m,1h),3.98(m,1h),3.66
–
3.53(m,3h),3.40(m,1h),3.23(m,1h),2.93(m,1h),2.47(s,3h).ms(esi):m/z 443.1[m h]
。
[0381]
实施例37化合物37的制备
[0382][0383]
将(3,5-二氟-4
–
((2-甲基)吡啶-4-基)氧基)苯基)甲醇6c(56mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物35e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物37(18mg,18.5%)。
[0384]1h nmr(400mhz,cdcl3)δ8.36(m,1h),7.09(m,2h),6.70
–
6.65(m,2h),5.70(s,1h),5.43(s,2h),4.41(m,1h),4.13(m,1h),4.04(m,1h),3.72
–
3.58(m,3h),3.46(m,1h),3.28(m,1h),2.96(m,1h),2.51(s,3h).ms(esi):m/z 443.1[m h]
。
[0385]
实施例38化合物38的制备
[0386][0387]
将(3,5-二氟-4-(3-(三氟甲基)苯氧基)苯基)甲醇10c(67mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物35e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体产物38(55mg,50.5%)。
[0388]1h nmr(400mhz,cdcl3)δ7.41(m,1h),7.33(m,1h),7.18(s,1h),7.10(m,3h),5.70(s,1h),5.41(s,2h),4.40(m,1h),4.15
–
4.07(m,1h),4.03(m,1h),3.70
–
3.55(m,3h),3.45(m,1h),3.27(m,1h),2.96(m,1h).ms(esi):m/z 496.1[m h]
。
[0389]
实施例39化合物39的制备
[0390][0391]
将3,5-二氟苯甲醇(32mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物35e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物39(37mg,50.2%)。
[0392]1h nmr(400mhz,cdcl3)δ6.96
–
6.89(m,2h),6.75(m,1h),5.68(s,1h),5.39(s,2h),4.40(m,1h),4.12(m,1h),4.03(m,1h),3.70
–
3.55(m,3h),3.44(m,1h),3.26(m,1h),2.95(m,1h).ms(esi):m/z 336.1[m h]
。
[0393]
实施例40化合物40的制备
[0394][0395]
将2,4,5-三氟苯甲醇(36mg,0.22mmol)溶于5ml干燥的n,n-二甲基甲酰胺中,0℃下加入氢化钠(矿物油中60%含量,11mg,0.26mmol),室温下搅拌反应5分钟后,加入化合物35e(50mg,0.22mmol),搅拌反应1小时后,加入少量水淬灭反应,用硅胶柱色谱法以洗脱剂体系(二氯甲烷/甲醇=20/1)纯化,得到白色固体目标产物40(39mg,50.2%)。
[0396]1h nmr(400mhz,cdcl3)δ7.36
–
7.28(m,1h),6.95(m,1h),5.65(s,1h),5.41(s,2h),4.42
–
4.35(m,1h),4.17
–
4.08(m,1h),4.03(m,1h),3.71
–
3.55(m,3h),3.44(m,1h),3.25(m,1h),2.95(m,1h).ms(esi):m/z 354.1[m h]
。
[0397]
生物学评价
[0398]
化合物的生物活性可通过使用用于测定作为lppla2抑制剂的化合物活性的任何适当测定方法及组织和体内模型来测定。
[0399]
(1)重组人lp-pla2测定(rhlp-pla2),也叫ped6的测定
[0400]
ped6是一种荧光标记的磷脂,可从invitogene或molecular probes直接购买。其sn3位上有荧光猝灭的对硝基苯基团,sn2位上有bodipy荧光素(fl)基团,一旦被lp-pla2酶断裂,释放出fl基团,导致荧光增强。但是lp-pla2抑制剂可以阻止这种断裂的产生,从而不会观察到荧光增强。
[0401]
测定方法:将待测化合物(如表1所示)与dmso溶液按照体积比1:3混合,经过稀释制成384孔微量板的源板。然后用echo液体分配器从源板上转移0.01微升化合物到384孔greiner784076板,将5微升由50mm hepes,ph7.4,150mm nacl,1mm chaps构成的缓冲液(该缓冲溶液中含有浓度为4nm或110pm的重组人lp-pla2酶)加到板上的各孔中。板以500rpm离心10秒钟,经过30分钟预温育后,将5微升上述缓冲液加到384孔greiner784076板,板以500rpm离心10秒钟,避光室温下温育20分钟后,用viewlux微量板成像仪在ex 480/em 540读取荧光强度,使用excel的xl拟合模型进行曲线和qc分析,计算pic50,结果列于表1。
[0402]
表1
[0403]
[0404]
(2)人血浆lp-pla2测定(又叫thio-paf测定)
[0405]
人血浆测定利用paf(磷脂酰胆碱)的硫脂类似物,它水解产生含自由巯基的磷脂,与cpm迈克尔加成生成增强荧光的马来酰亚胺,通过对荧光强度检测,可以对硫醇进行连续定量分析。该方法可以检测lp-pla2抑制剂对人血浆中lp-pla2酶的抑制活性。
[0406]
测定方法:将待测化合物(如表2所示)与dmso溶液按照体积比(1:3),经过稀释制成384孔的微量板的源板。用echo液体分配器从源板上转移0.01微升化合物到384孔greiner784076低容积板,然后加入预先等分并冷冻的8微升混合人血浆。板以500rpm离心10秒钟,经过30分钟预温育后,用bravo液体处理站将2微升的底物溶液,含2.5mm的2-硫代paf(乙醇溶液),32μm cpm(dmso溶液)和3.2mm n-乙基马来酰亚胺(nem)的缓冲液(50mm hepes,ph7.4,150mm nacl,1mm chaps构成的缓冲溶液)加到384孔greiner784076低容积板,2分钟后,用5微升5%的三氟乙酸猝灭反应,避光室温下温育40分钟后,用envision微量读板仪在ex 380/em 485读取荧光强度,使用excel的xl拟合模型进行曲线和qc分析,计算pic50,结果列于表2。
[0407]
表2
[0408]
[0409]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。