1.本发明属于医药领域,涉及一种苯并恶唑类小分子化合物、其制备方法及含有该化合物的药物组合物,及其在医药方面的应用。本发明公开了其作为zeste基因增强子同源物2(ezh2)抑制剂,用于预防和/或治疗与ezh2相关的疾病,如恶性肿瘤等。
背景技术:
2.恶性肿瘤是严重威胁人类健康的疾病,近年来其发病率和死亡率一直呈上升趋势,已成为全球面临的严峻的健康问题。肿瘤的发生发展是一个多因素多阶段的演进过程,涉及到多种基因的突变及表观遗传改变。表观遗传学是指在基因的dna序列不发生改变的情况下,基因的表达水平与功能发生改变,并产生可遗传表型的一种遗传学现象。多梳蛋白复合体(polycomb group protein,pcg)是参与染色质基因表观遗传负调控的重要蛋白因子,pcg家族包括多梳蛋白抑制性复合体1(prc1)和多梳蛋白抑制性复合体2(prc2)两种多聚复合物。zeste基因增强子同源物2(ezh2)为多梳蛋白复合体(pcg)家族的核心成员,ezh2是组成prc2蛋白复合物的一个催化亚基,在其功能中起核心作用。ezh2含有一个高度保守set结构域,具有组蛋白甲基转移酶(histone methyl transferase,hmt)活性,通过催化组蛋白的h3第27位赖氨酸三甲基化(h3k27me3)修饰,然后触发pcr1复合物成分在特定基因位点聚集从而导致下游靶基因沉默,二这些靶基因涉及了多种生物学基本过程的调控,如细胞凋亡、细胞周期调节、细胞老化和分化等。目前研究表明,ezh2高表达于多种肿瘤组织中,与肿瘤的恶性进展、侵袭性和转移能力密切相关。
3.ezh2的高表达常与人类癌症晚期的进展及不良预后有关,如:前列腺癌、乳腺癌、膀胱癌、肺癌、直肠癌、淋巴瘤等。ezh2的突变或缺失与肿瘤有关,如:弥漫性大b细胞淋巴瘤、滤泡性淋巴瘤、骨髓增生异常和骨髓增生性疾病等。目前,ezh2的y641和a677突变增加了编码的蛋白的活性,导致h3k27me3水平增高,从而促进了淋巴瘤细胞的增殖。
4.综上所述,ezh2作为一种表观遗传酶参与肿瘤的发生和发展,ezh2抑制剂作为药物在医药行业具有良好的应用前景。
5.已经公开的ezh2选择性抑制剂包括wo2012005805、wo2012050532、wo2012118812wo2015143424a2 wo2016102493a1 wo2017084494a1和wo2018045971a1等。目前已经公开了一系列的ezh2抑制剂专利,但仍需要开发新的ezh2抑制剂来满足市场的需求。
6.本发明重新设计合成了一类ezh2抑制剂,经实验研究,该类化合物对ezh2靶点的选择性高,体内动物实验能表现出优异的药效作用。
技术实现要素:
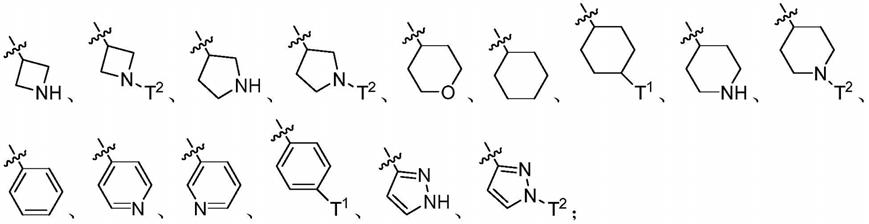
7.本发明的目的在于提供一种通式(i)所示的化合物,以及它们的互变异构体、对映体、非对映体、消旋体和可药用的盐。
[0008][0009]
r0为
‑
c1‑6烷基、
‑
c1‑3亚烷基
‑
nr
a
r
b
、
‑
t0或
‑
c1‑3亚烷基
‑
t0;
[0010]
r
a
和r
b
分别独立的为氢、
‑
c1‑3烷基、
‑
c1‑4亚烷基
‑
oh、
‑
t0、
‑
c1‑3亚烷基
‑
t0、
‑
(ch2)
n
‑
cf3、
‑
(ch2)
n
‑
chf2、
‑
(ch2)
n
‑
ch2f、
‑
c(o)
‑
c1‑3烷基、
‑
c(o)
‑
c2‑4烯基、
‑
c(o)
‑
(ch2)
n
‑
cf3、
‑
c(o)
‑
(ch2)
n
‑
chf2、
‑
c(o)
‑
(ch2)
n
‑
ch2f、
‑
c(o)
‑
t0、
‑
c(o)
‑
c1‑3亚烷基
‑
t0、
‑
c2‑4亚烷基
‑
och3或
‑
c2‑6亚烷基
‑
ch3,其中c2‑6亚烷基任选地被氧原子间断和/或任选地被一个或多个c1‑3烷基取代,r
a
和r
b
可进一步优选为甲基、乙基、丙基、
‑
c1‑4亚烷基
‑
oh、
‑
c2‑4亚烷基
‑
och3或
‑
c2‑6亚烷基
‑
ch3,其中c2‑6亚烷基任选地被氧原子间断;
[0011]
或是r
a
和r
b
连同它们所连接的氮原子一起形成未被取代或被取代的4
‑
6元杂环烷基,所述的4
‑
6元杂环烷上的亚甲基不被取代或是被1个或两个t基团取代,所述的4
‑
6元杂环烷基是杂原子为一个氮的杂环烷、杂原子为两个氮的杂环烷或是杂原子为一个氮和一个氧的杂环烷,当4
‑
6元杂环烷基是杂原子为两个氮的杂环烷时,环上的仲氮不被取代或是被t’取代;
[0012]
‑
nr
a
r
b
可进一步具体为可进一步具体为
[0013]
t为卤素、
‑
c1‑4烷基、被
‑
c1‑3烷基取代的
‑
c2‑4烷基或
‑
nr
c
r
d
,t可进一步优先为氟、
‑
c1‑4烷基、被
‑
c1‑2烷基取代的
‑
c2‑3烷基或
‑
nr
c
r
d
;
[0014]
当所述的4
‑
6元杂环烷上的一个亚甲基同时被两个t基团取代时,t为氟、甲基或乙基;
[0015]
r
c
和r
d
分别独立的为氢、
‑
c1‑3烷基、
‑
c1‑4亚烷基
‑
oh、
‑
c1‑4亚烷基
‑
och3或
‑
c2‑6亚烷基
‑
ch3,其中c2‑6亚烷基任选地被氧原子间断和/或任选地被一个或多个c1‑3烷基取代;
[0016]
t’为
‑
c1‑4烷基、被
‑
c1‑3烷基取代的
‑
c2‑4烷基、
‑
c1‑4亚烷基
‑
oh、
‑
c3‑6环烷基、4
‑
6元杂环烷基、
‑
(ch2)
n
‑
cf3、
‑
(ch2)
n
‑
chf2、
‑
(ch2)
n
‑
ch2f、
‑
c(o)
‑
c1‑3烷基、
‑
c(o)
‑
c2‑4烯基、
‑
c(o)
‑
(ch2)
n
‑
cf3、
‑
c(o)
‑
(ch2)
n
‑
chf2、
‑
c(o)
‑
(ch2)
n
‑
ch2f、叔丁氧羰基、
‑
s(o)2‑
c1‑3烷基、
‑
s(o)2‑
(ch2)
n
‑
cf3、
‑
s(o)2‑
(ch2)
n
‑
chf2;
[0017]
t0为未被取代或是被t1所取代的
‑
c3‑8环烷基、4
‑
6元杂环烷基、苯基或5
‑
6元杂芳基,当t0为4
‑
6元杂环烷基和5
‑
6元杂芳基时,若杂原子为氮原子,则氮原子不被取代或是被t2取代,t0进一步可具体为
[0018]
t1为卤素、
‑
c1‑6烷基、
‑
c1‑3烷氧基、被
‑
c1‑3烷基取代的
‑
c1‑6烷基或
‑
nr
c
r
d
;t1可进一步优先为氟、
‑
c1‑3烷基、
‑
c1‑3烷氧基、被
‑
c1‑2烷基取代的
‑
c2‑3烷基或
‑
nr
c
r
d
;
[0019]
t2为
‑
c1‑4烷基、被
‑
c1‑3烷基取代的
‑
c2‑4烷基、
‑
c1‑4亚烷基
‑
oh、
‑
c3‑6环烷基、4
‑
6元杂环烷基、
‑
(ch2)
n
‑
cf3、
‑
(ch2)
n
‑
chf2、
‑
(ch2)
n
‑
ch2f、
‑
c(o)
‑
c1‑3烷基、
‑
c(o)
‑
c2‑4烯基、
‑
c(o)
‑
(ch2)
n
‑
cf3、
‑
c(o)
‑
(ch2)
n
‑
chf2、
‑
c(o)
‑
(ch2)
n
‑
ch2f、叔丁氧羰基、
‑
s(o)2‑
c1‑3烷基、
‑
s(o)2‑
(ch2)
n
‑
cf3、
‑
s(o)2‑
(ch2)
n
‑
chf2;
[0020]
n为0、1、2、3或4;
[0021]
r3为氢、
‑
c1‑4烷基或被取代的
‑
c1‑4烷基,其中所述的被取代的
‑
c1‑4烷基任选被下列一个或多个取代基所取代:羟基、羧基或
‑
c(o)o
‑
r’,r3优选为甲基或乙基;
[0022]
r’为
‑
c1‑6烷基、
‑
c2‑6烯基、
‑
c2‑6炔基、
‑
c3‑8环烷基或
‑
c4‑
10
杂环烷基;
[0023]
r4和r5分别独立的为
‑
c1‑6烷基,优选为甲基或乙基;
[0024]
r
5a
为
‑
c1‑6烷基或
‑
c1‑6烷氧基,优选为甲基、乙基、甲氧基或乙氧基;
[0025]
r6为
‑
c1‑6烷基、五至六元环烷基、五至六元杂环烷基或具有8至10个碳原子的双环,所述的五至六元杂环烷基,杂原子选自一个氮、硫或氧,所述的双环是稠合相连的,双环中任何一个环是饱和的、不饱和的或是芳族的,所述的环烷基、杂环烷基或具有8至10个碳原子的双环不被取代或是被一个或多个r
6a
基团取代,r
6a
为卤素、羟基、
‑
c1‑3烷基、
‑
c1‑3烷氧基、3
‑
6元环烷基、4
‑
6元杂环基、
‑
nr
h
r
k
、
‑
c(o)
‑
c1‑3烷基、
‑
c(o)
‑
c3‑6环烷基、
‑
s(o)2‑
c1‑3烷基、
‑
s(o)2‑
c3‑6环烷基;
[0026]
当r6为含有一个硫原子的硫杂环基时,其中的硫杂原子不被氧化或被两个氧基氧化形成砜基;
[0027]
当r6为含有一个氮原子的氮杂环基时,其中的氮原子不被取代或是被r
6b
取代,r
6b
为
‑
c1‑4烷基、被
‑
c1‑3烷基取代的
‑
c2‑4烷基、
‑
c1‑4亚烷基
‑
oh、
‑
c3‑8环烷基、4
‑
6元杂环烷基、
‑
c(o)
‑
c1‑3烷基、
‑
c(o)
‑
c3‑6环烷基、
‑
s(o)2‑
c1‑3烷基、
‑
s(o)2‑
c3‑6环烷基,所述的4
‑
6元杂环烷基、杂原子选自氮或氧;
[0028]
r6优选为甲基、乙基、丙基、
[0029]
r
h
和r
k
分别独立的为氢、
‑
c1‑3烷基、
‑
c1‑4亚烷基
‑
oh、
‑
t0、
‑
c1‑3亚烷基
‑
t0、
‑
(ch2)
n
‑
cf3、
‑
(ch2)
n
‑
chf2、
‑
(ch2)
n
‑
ch2f、
‑
c(o)
‑
c1‑3烷基、
‑
c(o)
‑
c2‑4烯基、
‑
c(o)
‑
(ch2)
n
‑
cf3、
‑
c(o)
‑
(ch2)
n
‑
chf2、
‑
c(o)
‑
(ch2)
n
‑
ch2f、
‑
c(o)
‑
t0、
‑
c(o)
‑
c1‑3亚烷基
‑
t0、
‑
c2‑4亚烷基
‑
och3或
‑
c2‑6亚烷基
‑
ch3,其中c2‑6亚烷基任选地被氧原子间断和/或任选地被一个或多个c1‑3烷基取代,
[0030]
r
h
和r
k
或是连同它们所连接的氮原子一起形成未被取代或被1个或两个t基团取代的4
‑
6元杂环烷基,t为卤素、
‑
c1‑4烷基、被
‑
c1‑3烷基取代的
‑
c2‑4烷基或
‑
nr
c
r
d
,r
c
和r
d
分别独立的为氢、
‑
c1‑3烷基、
‑
c1‑4亚烷基
‑
oh、
‑
c2‑4亚烷基
‑
och3或
‑
c2‑6亚烷基
‑
ch3,其中c2‑6亚烷基任选地被氧原子间断和/或任选地被一个或多个c1‑3烷基取代。
[0031]
r
h
和r
k
优选为氢、
‑
c1‑3烷基、
‑
c2‑3亚烷基
‑
och3或
‑
c2‑6亚烷基
‑
ch3,其中,
[0032]
c2‑6亚烷基任选地被氧原子间断和/或任选地被一个或多个c1‑3烷基取代。
[0033]
本发明所提供化合物的合成流程:
[0034]
本发明的通式表示的化合物可以按照多种反应流程加以合成,本领域技术人员可以很容易的通过本文在实施例中所提供的一些制备方法设计其他化合物的反应流程。
[0035]
本发明涉及一种通式(i)所示化合物或其可药用的盐的制备方法,其中,当r0为
‑
c1‑3亚烷基
‑
nr
a
r
b
,m为1、2或3,通式(ii)所示化合物或其可药用的盐的制备方法,包括以下步骤:
[0036][0037]
通式(ii
‑
1)化合物在碱性条件下与二碳酸二叔丁酯发生反应得到通式(ii
‑
2)化合物;通式(ii
‑
2)化合物在钯炭还原剂条件下,被还原为通式(ii
‑
3)化合物;通式(ii
‑
3)化合物碱性条件下发生溴代反应,得到化合物为通式(ii
‑
4)化合物,该条件下提供的碱性条件的试剂优选为碳酸钙,溴化剂优选苄基三甲基三溴化铵;通式(ii
‑
4)化合物在碱性条件下,与氯乙酰氯发生反应,得到通式(ii
‑
5)化合物,该条件下提供的碱性条件的试剂优选为三乙胺;通式(ii
‑
5)化合物在碱性条件下,与相应的胺(r
a
r
b
nh)发生取代反应,得到通式(ii
‑
6)化合物,该条件下提供的碱性条件的试剂优选为碳酸铯;通式(ii
‑
6)化合物在加热,
碱性,催化剂存在的条件下,发生关环反应,得到通式(ii
‑
7)化合物,该条件下提供的碱性条件的试剂优选为碳酸铯,催化剂优选为碘化亚铜和1,10
‑
菲罗啉;通式(ii
‑
7)化合物在加热,碱性,催化剂存在的条件下,与相应的胺(r6‑
nh2)发生取代反应,得到通式(ii
‑
8)化合物,该条件下提供的碱性条件的试剂优选为碳酸铯,催化剂优选为三(二亚苄基丙酮)二钯和4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽;通式(ii
‑
8)化合物在还原剂(优选为乙酸硼氢化钠)的条件下,与相应的酮发生还原胺化反应得到通式(ii
‑
9)化合物;通式(ii
‑
9)化合物在酸性条件下,脱去叔丁基,得到通式(ii
‑
10)化合物,该条件下提供的酸性条件的试剂优选为三氟乙酸;通式(ii
‑
10)化合物与通式(ii
‑
11)化合物发生酰化反应,得到通式(i)化合物。
[0038]
当r0为
‑
c1‑6烷基、
‑
t0或
‑
c1‑3亚烷基
‑
t0,通式(i)所示化合物或其可药用的盐的制备方法,包括以下步骤:
[0039][0040]
通式(ii
‑
4)化合物在碱性条件下,与酰氯(r0cocl)发生反应,得到通式(iv
‑
1)化合物,该条件下提供的碱性条件的试剂优选为三乙胺;通式(iv
‑
1)化合物在加热,碱性,催化剂存在的条件下,发生关环反应,得到通式(iv
‑
2)化合物,该条件下提供的碱性条件的试剂优选为碳酸铯,催化剂优选为碘化亚铜和1,10
‑
菲罗啉;通式(iv
‑
2)化合物在加热,碱性,催化剂存在的条件下,与相应的胺(r6‑
nh2)发生取代反应,得到通式(iv
‑
3)化合物,该条件下提供的碱性条件的试剂优选为碳酸铯,催化剂优选为三(二亚苄基丙酮)二钯和4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽;通式(iv
‑
3)化合物在还原剂(优选为乙酸硼氢化钠)的条件下,与相应的醛或酮发生还原胺化反应得到通式(iv
‑
4)化合物;通式(iv
‑
4)化合物在酸性条件下,脱去叔丁基,得到通式(iv
‑
5)化合物,该条件下提供的酸性条件的试剂优选为三氟乙酸;通式(iv
‑
5)化合物与通式(ii
‑
11)化合物发生酰化反应,得到通式(iv)化合物。
[0041]
根据本发明通式(i)所示的化合物及其可药用的盐,其中该化合物具体为:
[0042]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0043]
n
‑
(4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)苯并[d]恶唑
‑4‑
甲酰胺;
[0044]2‑
环丙基
‑
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基苯并恶唑
‑4‑
甲酰胺;
[0045]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(1
‑
甲基
‑
1h
‑
吡唑
‑3‑
基)苯并[d]恶唑
‑4‑
甲酰胺;
[0046]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑
2,5
‑
二甲基苯并[d]恶唑
‑4‑
甲酰胺;
[0047]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑2‑
(4
‑
甲氧基苯基)
‑5‑
甲基苯并[d]恶唑
‑4‑
甲酰胺;
[0048]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吡啶
‑4‑
基)苯并[d]恶唑
‑4‑
甲酰胺;
[0049]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(甲基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0050]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
((二甲胺基)甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0051]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(哌啶
‑1‑
基甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0052]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
((4
‑
甲基哌嗪
‑1‑
基)甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0053]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
((4
‑
异丙基哌嗪
‑1‑
基)甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0054]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(((4
‑
二甲胺基)哌啶
‑1‑
基)甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0055]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0056]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(1
‑
甲基哌啶
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(二甲胺基甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0057]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
噻喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(二甲胺基甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0058]2‑
(氮杂环丁烷
‑1‑
基甲基)
‑
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基苯并[d]恶唑
‑4‑
甲酰胺;
[0059]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吡咯烷
‑1‑
基甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0060]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
噻喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
((4,4
‑
二氟哌啶
‑1‑
基)甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0061]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(4
‑
二甲氨基环己基)氨基)
‑5‑
甲基
‑2‑
((二甲氨基)甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0062]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(1
‑
乙酰基哌啶
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0063]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(1
‑
甲磺酰基哌啶
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0064]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(1
‑
甲基吡咯烷
‑3‑
基)氨基)
‑5‑
甲基
‑2‑
(二甲氨基)甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0065]6‑
((2,3
‑
二氢
‑
1h
‑
茚
‑2‑
基)(乙基)氨基)
‑
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡
啶
‑3‑
基)甲基)
‑2‑
(二甲氨基)甲基)
‑5‑
甲基
‑
苯并[d]恶唑
‑4‑
甲酰胺;
[0066]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(2
‑
吗啉代乙基)苯并[d]恶唑
‑4‑
甲酰胺;
[0067]
n
‑
((4
‑
甲氧基
‑6‑
甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺;
[0068]2‑
(二甲氨基甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑
n
‑
((4
‑
甲氧基
‑6‑
甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑5‑
甲基苯并[d]恶唑
‑4‑
甲酰胺;
[0069]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑2‑
(((2
‑
甲氧基乙基)(甲基)氨基)甲基)
‑5‑
甲基苯并[d]恶唑
‑4‑
甲酰胺;
[0070]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(1
‑
甲基哌啶
‑4‑
基)苯并[d]恶唑
‑4‑
甲酰胺;
[0071]
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(4
‑
甲氧基环己基)苯并[d]恶唑
‑4‑
甲酰胺。
[0072]
本发明所使用的术语“取代的”是指指定原子上的任何一个或多个氢原子被选自指定组的取代基取代,取代的结果是产生稳定的化合物,当取代基是氧代基团或酮基(即,=o)时,则原子上的2个氢原子被取代,酮取代基在芳香环上不存在。
[0073]
本发明所述的药用可接受的盐的是指无机碱盐,如钠盐、钾盐、钙盐、镁盐、锌盐、铵盐、季铵盐或铝盐;有机碱盐,如赖氨酸盐、精氨酸盐、二乙胺盐、三乙胺盐、乙醇胺盐、三甲胺盐、二环己基胺盐、胆碱盐、二苄胺盐、哌啶盐及其他药学上可接受的有机胺盐。
[0074]
在本发明的化合物分子中包含至少一个可成盐的氮原子时,可以通过在有机溶剂如乙腈、四氢呋喃中与相应的有机酸或无机酸反应,从而转化为相应的盐。典型的有机酸有草酸、酒石酸、马来酸、琥珀酸、甲磺酸、苯甲酸、苯磺酸、甲苯磺酸、氨基磺酸、柠檬酸、谷氨酸、焦谷氨酸、天冬氨酸、葡糖醛酸、萘磺酸、戊二酸、乙酸、三氟乙酸、苹果酸、富马酸、水杨酸、4
‑
氨基水杨酸、乳酸、棕榈酸盐、硬脂酸、月桂酸、肉桂酸、海藻酸、抗坏血酸盐,典型的无机酸有硝酸、盐酸、硫酸、磷酸。
[0075]
本发明的化合物中具有一个或多个不对称碳原子时,它们能够以如下形式存在:光学纯的对映异构体、纯的非对应异构体、对映异构体混合物、非对应异构体混合物、对映异构体外消旋混合物、外消旋物或外消旋物混合物。式(ii)的化合物的全部可能的异构体、立体异构体和其混合物也在本发明的范围内。
[0076]
本发明还提供了一种药物组合物,其包含上述至少一个化合物以及任选一种或多种医药上可接受的载剂和/或稀释剂。
[0077]
本发明所提供的药物组合物可以制备为任何形式,例如颗粒、粉末、片剂、包衣片剂、胶囊、药丸、糖浆、滴剂、溶液、混悬剂和乳剂,或者活性成分的缓释制剂,其中胶囊剂的实例包括硬或软明胶胶囊剂,颗粒剂和粉剂可以是非泡腾或泡腾形式。
[0078]
本发明的药物组合物可进一步包括一种或多种医药或生理上可接受的载体,这些载体将适当配制以便于给药。例如,医药或生理上可接受的载体可以是盐水、热压水、林格氏液、缓冲盐水、葡萄糖、麦芽糖糊精、甘油、乙醇及其混合物。本发明的药物组成物还可以包括医药或生理上可接受的添加剂,例如稀释剂、润滑剂、粘合剂、助流剂、崩解剂、甜味剂、矫味剂、湿润剂、分散剂、表面活性剂、溶剂、涂层剂、发泡剂、或芳香剂。
[0079]
可以使用的稀释剂的实例包括但不限于乳糖、蔗糖、淀粉、高岭土、盐、甘露糖醇和磷酸二钙;润滑剂的实例包括但不限于滑石、淀粉、镁或钙的硬脂酸盐、石松子和硬脂酸;粘合剂的实例包括但不限于微晶纤维素、黄蓍胶、葡萄糖溶液、阿拉伯胶浆、明胶溶液、蔗糖和淀粉糊;助流剂的实例包括但不限于胶体二氧化硅;崩解剂的实例包括但不限于交联羧甲基纤维素钠、淀粉羟乙酸钠、藻酸、玉米淀粉、马铃薯淀粉、膨润土、甲基纤维素、琼脂和羧甲基纤维素;甜味剂的实例包括但不限于蔗糖、乳糖、甘露糖醇和人工甜味剂,例如环磺酸钠和糖精,和任意数量的喷雾干燥矫味剂;矫味剂的实例包括但不限于从植物提取的天然矫味剂,例如果实,和味道较好的化合物,例如但不限于薄荷和水杨酸甲酯;湿润剂的实例包括但不限于丙二醇一硬脂酸酯、脱水山梨醇一油酸酯、二甘醇一月桂酸酯和聚氧乙烯月桂基醚。
[0080]
本发明的药物组合物可以根据传统方法来通过各种途径给药,包括口服、静脉内、动脉内、腹腔内、胸腔内、透皮、鼻腔、吸入、直肠、眼部和皮下导入。
[0081]
任选地添加到本发明的药物组合物中的医药上可接受的载体是:水、醇、蜂蜜、甘露醇、山梨醇、糊精、乳糖、焦糖、明胶、硫酸钙、硬脂酸镁、滑石粉、高岭土、甘油、吐温、琼脂、碳酸钙、碳酸氢钙、表面活性剂、环糊精及其衍生物、磷脂类、磷酸盐类、淀粉类及其衍生物、硅衍生物、纤维素类及其衍生物、吡咯烷酮类、聚乙二醇类、丙烯酸树脂类、酞酸酯类、丙烯酸共聚物、苯三酸酯类中的一种或几种。
[0082]
经药理实验验证,本发明所提供的化合物或者药物组合物可通过ezh2治疗肿瘤、骨髓增生性疾病或自身免疫性疾病,所述的肿瘤为淋巴瘤、黑色素瘤、神经胶质瘤、胃肠间质瘤、前列腺癌、乳腺癌、卵巢癌、膀胱癌、肺癌、直肠癌、皮肤癌、上皮细胞癌、鼻咽癌、骨癌、食道癌或白血病,所述的自身免疫性疾病为炎症性肠炎、自身免疫性脑脊髓炎或多发性硬化。
[0083]
本发明所提供的化合物一般的剂量范围为约每天0.001mg/kg至1000mg/kg,优选为约0.01mg/kg至100mg/kg,更优选为约0.1至20mg/kg,药物组合物的剂量范围为以其含有的上述化合物的量来计算。
具体实施方式
[0084]
以下结合实施例进一步描述本发明,但这些实施例并非限制着本发明的范围。
[0085]
实施例1n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0086][0087]
第一步2
‑
甲基
‑6‑
硝基苯甲酸叔丁酯1b
[0088]
化合物2
‑
甲基
‑6‑
硝基苯甲酸1a(4.0g,22.1mmol)溶于四氢呋喃中(40ml),加入二碳酸二叔丁酯(12g,55.2mmol)和4
‑
二甲氨基吡啶(2.7g,22.1mmol),室温下搅拌过夜。加入水和乙酸乙酯萃取,有机相用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后柱层析纯化得到标题化合物1b(1.8g,7.6mmol),收率34%。
[0089]
第二步2
‑
氨基
‑6‑
甲基苯甲酸叔丁基酯1c
[0090]
化合物1b(1.8g,7.6mmol)溶于甲醇(20ml),加入钯碳(0.9g),在氢气保护下室温搅拌2h,tlc检测原料反应完毕,过滤,浓缩后得到标题化合物1c(1.5g,7.2mmol),收率95%。
[0091]
第三步2
‑
氨基
‑
3,5
‑
二溴
‑6‑
甲基苯甲酸叔丁酯1d
[0092]
化合物1c(1.5g,7.25mmol)溶于二氯甲烷(30ml),加入苄基三甲基三溴化铵(7.0g,18mmol)和碳酸钙(1.9g,18mmol),室温下搅拌过夜。反应完毕,过滤后加水和乙酸乙酯萃取,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后柱层析纯化得到标题化合物1d(2.4g,6.6mmol),收率91%。
[0093]
第四步3,5
‑
二溴
‑2‑
(2
‑
氯乙酰氨基)
‑6‑
甲基苯甲酸叔丁酯1e
[0094]
化合物1d(1.7g,4.66mmol)溶于二氯甲烷(20ml),加入三乙胺(1.4g,14mmol),再慢慢滴加氯乙酰氯(1.6g,14mmol),室温下搅拌4h。反应完毕后,加水和乙酸乙酯萃取,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后柱层析纯化得到标题化合物1e(1.6g,3.6mmol),收率78%。
[0095]
第五步3,5
‑
二溴
‑2‑
甲基
‑6‑
(2
‑
吗啉代乙酰胺基)苯甲酸叔丁酯1f
[0096]
化合物1e(0.8g,1.8mmol)溶于n,n
‑
二甲基甲酰胺(10ml),加入吗啉(0.32g,3.6mmol和碳酸铯(0.6g,1.8mmol),室温下搅拌2h。反应完毕后,加水和乙酸乙酯萃取,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后柱层析纯化得到标题化合物1f(0.6g,1.2mmol),收率67%。
[0097]
第六步6
‑
溴
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酸叔丁酯1g
[0098]
化合物1f(550mg,1.12mmol)和碘化亚铜(107mg,0.56mmol)溶于乙二醇二甲醚(10ml),加入化合物1,10
‑
菲罗啉(162mg,0.44mmol)和碳酸铯(730mg,2.24mmol),在氮气保护下加热110℃下搅拌3h。加入乙酸乙酯和水,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后柱层析纯化得到标题化合物1g(300mg,0.73mmol),收率65%。
[0099]1h nmr(400mhz,dmso)δ8.19(s,1h),3.83(s,2h),3.56
–
3.53(t,j=4.4hz,4h),2.51
‑
2.38(m,4h),2.38(s,3h),1.54(s,9h)。
[0100]
第七步5
‑
甲基
‑2‑
(吗啉代甲基)
‑6‑
((四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)苯并[d]恶唑
‑4‑
甲酸叔丁酯1h
[0101]
化合物1g(300mg,0.73mmol)和四氢
‑
2h
‑
吡喃
‑4‑
胺(147mg,1.46mmol)溶于甲苯(10ml),加入无水碳酸铯(476mg,1.46mmol),三(二亚苄基丙酮)二钯(67mg,0.073mmol),4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽(43mg,0.073mmol),在氮气保护下加热100℃下搅拌3h。冷却后,旋干,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物1h(150mg,0.35mmol),收率47.6%。
[0102]
ms m/z(esi):432[m h]
。
[0103]
第八步6
‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
羧酸叔丁酯1i
[0104]
化合物1h(70mg,0.16mmol)溶于1,2
‑
二氯乙烷(5.0ml),加入乙酸(58mg,0.96mmol)和乙醛(22mg,0.48mmol),反应在常温下搅拌30分钟后,再加入醋酸硼氢化钠(103mg,0.48mmol),在常温下搅拌2h后,反应完毕。反应液用水淬灭,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物1i(50mg,0.11mmol),收率67%。
[0105]
第九步6
‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
羧酸1j
[0106]
化合物1i(50mg,0.11mmol)溶于二氯甲烷(2.0ml),加入三氟乙酸(1.0ml),反应在常温下搅拌2h后,反应完毕。反应液用水洗,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物1j(40mg,0.10mmol),收率91%。
[0107]
第十步n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0108]
化合物1j(40mg,0.10mmol)和3
‑
(氨甲基)
‑
4,6
‑
二甲基
‑
1h
‑
吡啶
‑2‑
酮盐酸盐(22mg,0.12mmol)溶于n,n
‑
二甲基甲酰胺(5.0ml),加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(38mg,0.2mmol),1
‑
羟基苯并三唑(14mg,0.1mmol)和三乙胺(50mg,0.5mmol)反应在常温下搅拌2h后,反应完毕。反应液用水洗,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物1(20mg,0.037mmol),收率38%。
[0109]1h nmr(400mhz,dmso)δ11.42(s,1h),8.24(s,1h),7.53(s,1h),5.83(s,1h),4.32
(d,j=4.8hz,1h)3.80
‑
3.76(d,j=16hz,4h),3.55
‑
3.52(t,j=4.4hz,4h),3.21
‑
3.16(t,j=11.2hz,2h),3.02
‑
2.99(t,j=7.6hz,2h),2.91(m,1h),2.21(s,6h),2.06(s,3h),1.61
‑
1.58(d,j=12hz,2h),1.44(m,2h),0.77
‑
0.73(t,j=7.2hz,3h)。
[0110]
ms m/z(esi):538[m h]
。
[0111]
实施例2n
‑
(4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)苯并[d]恶唑
‑4‑
甲酰胺
[0112][0113]
第一步3,5
‑
二溴
‑2‑
甲基
‑6‑
(四氢
‑
2h
‑
吡喃
‑4‑
甲酰胺基)苯甲酸叔丁酯5a
[0114]
化合物1d(530mg,1.452mmol)溶于二氯甲烷(20ml),加入三乙胺(147mg,1.452mmol),再慢慢滴加四氢
‑
2h
‑
吡喃
‑4‑
甲酰氯(644mg,4.356mmol),室温下搅拌4h。反应完毕后,加水和乙酸乙酯萃取,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后柱层析纯化得到标题化合物5a(480mg,1.01mmol),收率69%。
[0115]
第二步6
‑
溴
‑5‑
甲基
‑2‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)苯并[d]恶唑
‑4‑
羧酸叔丁酯5b
[0116]
化合物5a(480mg,1.01mmol)和碘化亚铜(190mg,1mmol)溶于乙二醇二甲醚(10ml),加入化合物1,10
‑
菲罗啉(180mg,1mmol)和碳酸铯(652mg,2mmol),在氮气保护下加热110℃下搅拌3h。加入乙酸乙酯和水,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后柱层析纯化得到标题化合物5b(260mg,0.658mmol),收率65%。
[0117]
第三步5
‑
甲基
‑2‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)
‑6‑
((四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)苯并[d]恶唑
‑4‑
羧酸叔丁酯5c
[0118]
化合物5b(260mg,0.658mmol)和四氢
‑
2h
‑
吡喃
‑4‑
胺(133mg,1.316mmol)溶于甲苯(10ml),加入无水碳酸铯(429mg,1.316mmol),三(二亚苄基丙酮)二钯(60mg,0.0658mmol),4,5
‑
双二苯基膦
‑
9,9
‑
二甲基氧杂蒽(76mg,0.1316mmol),在氮气保护下加热100℃下搅拌3h。冷却后,旋干,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物5c(130mg,0.3125mmol),收率48%。
[0119]
ms m/z(esi):417[m h]
。
[0120]
第四步6
‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)苯
并[d]恶唑
‑4‑
羧酸叔丁酯5d
[0121]
化合物5c(130mg,0.3125mmol)溶于1,2
‑
二氯乙烷(5.0ml),加入乙酸(19mg,0.3125mmol)和乙醛(28mg,0.625mmol),反应在常温下搅拌30分钟后,再加入醋酸硼氢化钠(133mg,0.625mmol),在常温下搅拌2h后,反应完毕。反应液用水淬灭,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物5d(90mg,0.202mmol),收率64.6%。
[0122]
第五步6
‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)苯并[d]恶唑
‑4‑
羧酸5e
[0123]
化合物5d(90mg,0.202mmol)溶于二氯甲烷(2.0ml),加入三氟乙酸(1.0ml),反应在常温下搅拌2h后,反应完毕。反应液用水洗,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物5e(77mg,0.2mmol),收率99%。第六步n
‑
(4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(四氢
‑
2h
‑
吡喃
‑4‑
基)苯并[d]恶唑
‑4‑
甲酰胺5
[0124]
化合物5e(77mg,0.2mmol)和3
‑
(氨甲基)
‑
4,6
‑
二甲基
‑
1h
‑
吡啶
‑2‑
酮盐酸盐(87mg,0.46mmol)溶于n,n
‑
二甲基甲酰胺(5.0ml),加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(88mg,0.46mmol),1
‑
羟基苯并三唑(63mg,0.46mmol)和三乙胺(187mg,1.85mmol)反应在常温下搅拌2h后,反应完毕。反应液用水洗,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物5(38mg,0.072mmol),收率36%。
[0125]1h nmr(400mhz,dmso
‑
d6)δ11.45(s,1h),8.27(t,j=5.1hz,1h),7.52(s,1h),5.84(s,1h),4.32(d,j=5.0hz,2h),3.87(d,j=11.1hz,2h),3.78(d,j=11.3hz,2h),3.52
–
3.40(m,2h),3.28
–
3.13(m,3h),3.06
–
2.85(m,3h),2.24(d,j=5.4hz,5h),2.07(s,3h),1.95(d,j=13.0hz,2h),1.77(dd,j=12.8,8.6hz,2h),1.60(d,j=12.4hz,2h),1.44(d,j=11.1hz,2h),0.74(t,j=6.9hz,3h).
[0126]
ms m/z(esi):523[m h]
。
[0127]
实施例3 2
‑
环丙基
‑
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基苯并恶唑
‑4‑
甲酰胺
[0128][0129]
采用与实施例2类似的合成方法,将四氢
‑
2h
‑
吡喃
‑4‑
甲酰氯替换为环丙基甲酰氯,制得标题产物6,收率33%。
[0130]1h nmr(400mhz,dmso
‑
d6)δ11.46(s,1h),7.44(s,1h),5.84(s,1h),4.31(d,j=5.1hz,2h),3.30(s,1h),3.18(td,j=13.5,12.6,2.9hz,2h),2.99(q,j=6.9hz,2h),2.23(d,j=5.9hz,6h),2.07(s,3h),1.59(d,j=12.5hz,2h),1.48
–
1.36(m,1h),1.11(dt,j=8.2,3.1hz,2h),1.06(dt,j=5.3,2.9hz,2h),0.73(t,j=6.9hz,3h).
[0131]
ms m/z(esi):479[m h]
。
[0132]
实施例4n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(1
‑
甲基
‑
1h
‑
吡唑
‑3‑
基)苯并[d]恶唑
‑4‑
甲酰胺
[0133][0134]
采用与实施例2类似的合成方法,将四氢
‑
2h
‑
吡喃
‑4‑
甲酰氯替换为1
‑
甲基
‑
1h
‑
吡唑
‑3‑
基甲酰氯,制得标题产物7,收率13%。
[0135]1h nmr(400mhz,dmso
‑
d6)δ11.47(s,1h),8.37(t,j=5.0hz,1h),7.91(d,j=2.3hz,1h),7.57(s,1h),6.88(d,j=2.3hz,1h),5.85(s,1h),4.35(d,j=4.9hz,2h),3.95(s,3h),3.84
–
3.71(m,2h),3.20(t,j=11.4hz,2h),3.09
–
2.88(m,3h),2.25(d,j=4.0hz,6h),2.07(s,3h),1.62(d,j=12.4hz,2h),1.48(dt,j=12.0,5.9hz,2h),0.77(t,j=6.9hz,3h).
[0136]
ms m/z(esi):519[m h] 。
[0137]
实施例5n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑
2,5
‑
二甲基苯并[d]恶唑
‑4‑
甲酰胺8
[0138][0139]
采用与实施例2类似的合成方法,将四氢
‑
2h
‑
吡喃
‑4‑
甲酰氯替换为乙酰氯,制得标题产物8,收率40%。
[0140]1h nmr(400mhz,chloroform
‑
d)δ11.16(s,1h),7.98(s,1h),7.26(s,1h),5.94(s,1h),4.63(d,j=5.8hz,2h),3.92(d,j=11.4hz,2h),3.28(td,j=11.2,3.7hz,2h),3.06(d,j=7.2hz,2h),2.94(s,1h),2.54(s,3h),2.47(s,2h),2.44(s,3h),2.22
–
2.17(m,3h),1.66(s,2h),1.23(s,3h),0.84(t,j=7.0hz,3h).
[0141]
ms m/z(esi):453.1[m h]
[0142]
实施例6n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑2‑
(4
‑
甲氧基苯基)
‑5‑
甲基苯并[d]恶唑
‑4‑
甲酰胺9
[0143][0144]
采用与实施例2类似的合成方法,将四氢
‑
2h
‑
吡喃
‑4‑
甲酰氯替换为4
‑
甲氧基苯甲酰氯,制得标题产物9,收率9%。
[0145]
ms m/z(esi):545[m h]
[0146]
实施例7n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吡啶
‑4‑
基)苯并[d]恶唑
‑4‑
甲酰胺10
[0147][0148]
采用与实施例2类似的合成方法,将四氢
‑
2h
‑
吡喃
‑4‑
甲酰氯替换为吡啶
‑4‑
甲酰氯,制得标题产物10,收率8%。
[0149]
ms m/z(esi):516[m h]
[0150]
实施例8n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(甲基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0151][0152]
第一步5
‑
甲基
‑6‑
(甲基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
羧酸叔丁酯14a
[0153]
化合物1h(70mg,0.16mmol)溶于1,2
‑
二氯乙烷(5.0ml),加入乙酸(58mg,0.96mmol)和多聚甲醛(31mg,0.98mmol),反应在常温下搅拌30分钟后,再加入醋酸硼氢化钠(103mg,0.48mmol),在常温下搅拌2h后,反应完毕。反应液用水淬灭,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物14a(80mg,0.16mmol),收率100%。
[0154]
第二步5
‑
甲基
‑6‑
(甲基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
羧酸14b
[0155]
化合物14a(80mg,0.16mmol)溶于二氯甲烷(2.0ml),加入三氟乙酸(1.0ml),反应在常温下搅拌2h后,反应完毕。反应液用水洗,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物14b(54mg,0.14mmol),收率88%。
[0156]
第三步n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(甲基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺14
[0157]
化合物14b(54mg,0.14mmol)和3
‑
(氨甲基)
‑
4,6
‑
二甲基
‑
1h
‑
吡啶
‑2‑
酮盐酸盐
(39mg,0.21mmol)溶于n,n
‑
二甲基甲酰胺(5.0ml),加入1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(54mg,0.28mmol),1
‑
羟基苯并三唑(19mg,0.14mmol)和三乙胺(43mg,0.42mmol)反应在常温下搅拌2h后,反应完毕。反应液用水洗,用二氯甲烷萃取三次,用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩后板层析纯化得到标题化合物14(33.2mg,0.063mmol),收率45%。
[0158]1h nmr(400mhz,dmso
‑
d6)δ11.43(s,1h),8.23(t,j=5.0hz,1h),7.49(s,1h),5.83(s,2h),4.31(d,j=5.1hz,2h),3.80(d,j=11.2hz,2h),3.76(s,2h),3.54(t,j=4.6hz,4h),3.26
–
3.16(m,2h),2.93(dd,j=10.5,5.6hz,1h),2.56(s,3h),2.47(d,j=1.9hz,4h),2.22(s,6h),2.07(d,j=0.9hz,3h),1.60
–
1.48(m,4h).
[0159]
ms m/z(esi):524.3[m h] 。
[0160]
实施例9n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
((二甲胺基)甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0161][0162]
采用与实施例1类似的合成方法,将吗啉替换为二甲胺,制得标题产物15,收率22%。
[0163]
ms m/z(esi):496[m h]
。
[0164]
实施例10n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(哌啶
‑1‑
基甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0165][0166]
采用与实施例1类似的合成方法,将吗啉替换为哌啶,制得标题产物30,收率10%。
[0167]
ms m/z(esi):536.3[m h]
。
[0168]
实施例11n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
((4
‑
甲基哌嗪
‑1‑
基)甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0169]
[0170]
采用与实施例1类似的合成方法,将吗啉替换为1
‑
甲基哌嗪,制得标题产物31,收率15%。
[0171]
ms m/z(esi):551.3[m h]
。
[0172]
实施例12n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
((4
‑
异丙基哌嗪
‑1‑
基)甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0173][0174]
采用与实施例1类似的合成方法,将吗啉替换为1
‑
异丙基哌嗪,制得标题产物31,收率15%。
[0175]
ms m/z(esi):579.3[m h]
。
[0176]
实施例13n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(((4
‑
二甲胺基)哌啶
‑1‑
基)甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0177][0178]
采用与实施例1类似的合成方法,将吗啉替换为4
‑
二甲胺基哌啶,制得标题产物33,收率26%。
[0179]
ms m/z(esi):579.3[m h]
。
[0180]
实施例14n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0181][0182]
采用与实施例1类似的合成方法,将乙醛替换为多聚甲醛,制得标题产物34,收率13%。
[0183]
ms m/z(esi):524.2[m h]
。
[0184]
实施例15n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(1
‑
甲基哌啶
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(二甲胺基甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0185][0186]
采用与实施例1类似的合成方法,将吗啉替换为二甲胺,将四氢吡喃酮替换为n
‑
甲基
‑4‑
哌啶酮,制得标题产物35,收率9%。
[0187]
ms m/z(esi):509[m h]
。
[0188]
实施例16n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
噻喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(二甲胺基甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0189][0190]
采用与实施例1类似的合成方法,将吗啉替换为二甲胺,将四氢吡喃酮替换为四氢噻喃酮,制得标题产物36,收率11%。
[0191]
ms m/z(esi):512[m h] 。
[0192]
实施例17 2
‑
(氮杂环丁烷
‑1‑
基甲基)
‑
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基苯并[d]恶唑
‑4‑
甲酰胺
[0193][0194]
采用与实施例1类似的合成方法,将吗啉替换为氮杂环丁烷,制得标题产物37,收率26%。
[0195]
ms m/z(esi):508[m h] 。
[0196]
实施例18n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吡咯烷
‑1‑
基甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0197][0198]
采用与实施例1类似的合成方法,将吗啉替换为四氢吡咯,制得标题产物38,收率8%。
[0199]
ms m/z(esi):522.3[m h] 。
[0200]
实施例19n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
噻喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
((4,4
‑
二氟哌啶
‑1‑
基)甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0201][0202]
采用与实施例1类似的合成方法,将吗啉替换为四氢吡咯,制得标题产物39,收率35%。
[0203]
ms m/z(esi):572.3[m h] 。
[0204]
实施例20n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(4
‑
二甲氨基环己基)氨基)
‑5‑
甲基
‑2‑
((二甲氨基)甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0205][0206]
采用与实施例1类似的合成方法,将吗啉替换为二甲胺,将四氢吡喃酮替换为4
‑
二甲氨基环己酮,制得标题产物40,收率18%。
[0207]
ms m/z(esi):537.3[m h] 。
[0208]
实施例21n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(1
‑
乙酰基哌啶
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0209][0210]
采用与实施例1类似的合成方法,将将四氢吡喃酮替换为1
‑
乙酰基哌啶
‑4‑
酮,制得标题产物41,收率17%。
[0211]
ms m/z(esi):579.3[m h] 。
[0212]
实施例22n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(1
‑
甲磺酰基哌啶
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0213][0214]
采用与实施例1类似的合成方法,将四氢吡喃酮替换为1
‑
甲磺酰基哌啶
‑4‑
酮,制得标题产物42,收率35%。
[0215]
ms m/z(esi):615[m h] 。
[0216]
实施例23n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(1
‑
甲基吡咯烷
‑3‑
基)氨基)
‑5‑
甲基
‑2‑
(二甲氨基)甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0217][0218]
采用与实施例1类似的合成方法,将吗啉替换为二甲胺,将四氢吡喃酮替换为1
‑
甲基
‑3‑
吡咯烷酮,制得标题产物43,收率29%。
[0219]
ms m/z(esi):495[m h] 。
[0220]
实施例24 6
‑
((2,3
‑
二氢
‑
1h
‑
茚
‑2‑
基)(乙基)氨基)
‑
n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑2‑
(二甲氨基)甲基)
‑5‑
甲基
‑
苯并[d]恶唑
‑4‑
甲酰胺
[0221][0222]
采用与实施例1类似的合成方法,将吗啉替换为二甲胺,将四氢吡喃酮替换为2
‑
茚酮,制得标题产物44,收率18%。
[0223]
ms m/z(esi):528[m h] 。
[0224]
实施例25n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(2
‑
吗啉代乙基)苯并[d]恶唑
‑4‑
甲酰胺
[0225][0226]
采用与实施例1类似的合成方法,将氯乙酰氯换为3
‑
氯丙酰氯,制得标题产物45,收率38%。
[0227]
ms m/z(esi):552.3[m h] 。
[0228]
实施例26n
‑
((4
‑
甲氧基
‑6‑
甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(吗啉代甲基)苯并[d]恶唑
‑4‑
甲酰胺
[0229][0230]
采用与实施例1类似的合成方法,将3
‑
(氨甲基)
‑
4,6
‑
二甲基
‑
1h
‑
吡啶
‑2‑
酮盐酸
盐换为3
‑
(氨甲基)
‑4‑
甲氧基
‑6‑
甲基
‑
1h
‑
吡啶
‑2‑
酮盐酸盐,制得标题产物46,收率10%。
[0231]
ms m/z(esi):554.3[m h] 。
[0232]
实施例27 2
‑
(二甲氨基甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑
n
‑
((4
‑
甲氧基
‑6‑
甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑5‑
甲基苯并[d]恶唑
‑4‑
甲酰胺
[0233][0234]
采用与实施例1类似的合成方法,将吗啉替换为二甲胺,将3
‑
(氨甲基)
‑
4,6
‑
二甲基
‑
1h
‑
吡啶
‑2‑
酮盐酸盐换为3
‑
(氨甲基)
‑4‑
甲氧基
‑6‑
甲基
‑
1h
‑
吡啶
‑2‑
酮盐酸盐,制得标题产物47,收率29%。
[0235]
ms m/z(esi):512[m h] 。
[0236]
实施例28n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑2‑
(((2
‑
甲氧基乙基)(甲基)氨基)甲基)
‑5‑
甲基苯并[d]恶唑
‑4‑
甲酰胺
[0237][0238]
采用与实施例1类似的合成方法,将吗啉替换为2
‑
甲氧基
‑
n
‑
甲基乙胺,制得标题产物48,收率18%。
[0239]
ms m/z(esi):540.3[m h] 。
[0240]
实施例29n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(1
‑
甲基哌啶
‑4‑
基)苯并[d]恶唑
‑4‑
甲酰胺
[0241][0242]
采用与实施例2类似的合成方法,将四氢
‑
2h
‑
吡喃
‑4‑
甲酰氯替换为1
‑
甲基基哌啶
‑4‑
甲酰氯,制得标题产物79,收率20%。
[0243]
ms m/z(esi):536.2[m h] 。
[0244]
实施例30n
‑
((4,6
‑
二甲基
‑2‑
氧代
‑
1,2
‑
二氢吡啶
‑3‑
基)甲基)
‑6‑
(乙基(四氢
‑
2h
‑
吡喃
‑4‑
基)氨基)
‑5‑
甲基
‑2‑
(4
‑
甲氧基环己基)苯并[d]恶唑
‑4‑
甲酰胺
[0245][0246]
采用与实施例2类似的合成方法,将四氢
‑
2h
‑
吡喃
‑4‑
甲酰氯替换为4
‑
甲氧基环己酰氯,制得标题产物80,收率11%。
[0247]
ms m/z(esi):551[m h] 。
[0248]
生物学实验
[0249]
实验一、本发明化合物对野生型多梳蛋白复合物2(polycomb repressive complex 2,prc2)(果蝇泽斯特基因增强子同源物2(enhancer of zeste homolog 2,ezh2)野生型)活性的测定
[0250]
一、检测方法同位素法(radiometric assay)
[0251]
二、实验步骤
[0252]
每孔加入待测化合物、10微升(μl)野生型ezh2,室温孵育15分钟(min)后,加入多肽和[3h]标记的甲基供体s
‑
腺苷甲硫氨酸(s
‑
adenosylmethionine,sam)室温反应1小时(h),加入冷的sam终止反应。转移25μl反应液到闪烁板中,室温孵育1h后用去离子水和0.1%吐温20洗板三次,用珀金埃尔默(perkinelmer)液体闪烁/发光计数仪读值,用棱镜5软件(graphpad prism 5)计算化合物的半数抑制浓度(ic50)。
[0253]
三、实验结果
[0254]
结果显示,阳性药epz
‑
6438在对野生型ezh2抑制的ic50为5.1纳摩尔每升(nm),实施例化合物野生型ezh2均有较强的抑制作用,结果见表1。
[0255]
实验二、本发明化合物对突变型prc2复合物(ezh2突变型)活性的测定
[0256]
一、检测方法均相时间分辨荧光法(homogeneous time
‑
resolved fluorescence,htrf)
[0257]
二、实验步骤
[0258]
每孔加入prc2复合物(ezh2 y641f/a677g/y641n)、h3(1
‑
50)me1底物、甲基供体sam及化合物,总反应体系为10微升(μl),室温避光反应4小时。每孔加入5微升(μl)eu标记的h3k27 me3抗体和5微升(μl)。
[0259]
三、实验结果
[0260]
结果显示,阳性药epz
‑
6438在对突变型y641f ezh2抑制的ic50为4.6nm,a677g ezh2抑制的ic50为2.91nm,y641n ezh2抑制的ic50为2.24nm,实施例化合物对突变型ezh2均有较强的抑制作用,结果见表1。
[0261]
表1.化合物对prc2复合物的抑制活性
[0262][0263]
[0264]
实验三、本发明化合物对弥散性巨型b细胞淋巴瘤细胞(wsu
‑
dlcl2细胞)增殖分析一、实验步骤
[0265]
弥散性巨型b细胞淋巴瘤细胞(wsu
‑
dlcl2细胞系)生长状态良好时,收集细胞,计数。调整细胞浓度至100000个细胞/每毫升(cells/ml)。将以上浓度的细胞接种与96孔板中,100微升(μl)/孔(每孔内细胞数量为10000)。在二甲基亚砜(dmso)中梯度稀释化合物,配制500倍终浓度的化合物板。吸取1.2微升(μl)500x的化合物,转入200微升(μl)的培养基中,吹打混匀,得到3x的化合物中间板。吸取50微升(μl)3x的化合物,按照设定的排布加入细胞板。将细胞板置于二氧化碳培养箱培养4天。取出细胞板,混匀,吸取一定体积的细胞悬液,以钙黄绿素am(calcein am)染色,艾库门(acumen)计数每孔的细胞数量。根据检测的细胞数量,重新接种10000细胞于96孔板中。同上加入化合物。置于二氧化碳培养箱培养3天(第7天)。再次取出细胞板,混匀,吸取一定体积的细胞悬液,以钙黄绿素am(calcein am)染色,艾库门(acumen)计数每孔的细胞数量。继续培养细胞4天(第11天),第三次取出细胞板,混匀,吸取一定体积的细胞悬液,以钙黄绿素am(calcein am)染色,艾库门(acumen)计数每孔的细胞数量。根据检测的细胞数量,重新接种10000细胞于96孔板中。同上加入化合物。置于二氧化碳培养箱培养3天(第7天)。继续培养细胞3天(第14天),重复步骤5,得到最终计数结果。处理day 14的数据,得到相应的半数抑制浓度ic50。
[0266]
二、实验结果及结论
[0267]
结果显示,epz
‑
6438在对wsu
‑
dlcl2增殖抑制的ic50为67.23nm,实施例中化合物wsu
‑
dlcl2增殖均有较强的抑制作用,抑制活性优于epz6438。结果见表2。
[0268]
实验四、本发明化合物对人弥漫性大细胞淋巴瘤b淋巴细胞(pfeiffer细胞)增殖分析一、实验步骤
[0269]
人弥漫性大细胞淋巴瘤b淋巴细胞(pfeiffer)生长状态良好时,收集细胞,计数。调整细胞浓度至100000个/每毫升。将以上浓度的细胞接种与96孔板中,100微升(μl)/孔(每孔内细胞数量为10000)。在dmso中梯度稀释化合物,配制500倍终浓度的化合物板。吸取1.2微升(μl)500x的化合物,转入200μl的培养基中,吹打混匀,得到3x的化合物中间板。吸取50微升(μl)3x的化合物,按照设定的排布加入细胞板。将细胞板置于二氧化碳培养箱培养4天。取出细胞板,混匀,吸取一定体积的细胞悬液,以钙黄绿素am(calcein am)染色,艾库门(acumen)计数每孔的细胞数量。根据检测的细胞数量,重新接种10000细胞于96孔板中。同上加入化合物。置于二氧化碳培养箱培养3天(第7天)。再次取出细胞板,混匀,吸取一定体积的细胞悬液,以钙黄绿素am(calcein am)染色,艾库门(acumen)计数每孔的细胞数量。继续培养细胞4天(第11天),第三次取出细胞板,混匀,吸取一定体积的细胞悬液,以钙黄绿素am(calcein am)染色,艾库门(acumen)计数每孔的细胞数量。6.根据检测的细胞数量,重新接种10000细胞于96孔板中。同上加入化合物。置于二氧化碳培养箱培养3天(第7天)。继续培养细胞3天(第14天),重复步骤5,得到最终计数结果。处理day 14的数据,得到相应的半数抑制浓度ic50。
[0270]
二、实验结果及结论
[0271]
结果显示,epz
‑
6438在对pfeiffer增殖抑制的ic50为27.14nm,实施例化合物对pfeiffer细胞增殖均有较强的抑制作用,抑制活性优于epz6438。结果见表2。
[0272]
表2.化合物对ezh2敏感细胞增殖的抑制作用
[0273][0274][0275]
实验五、本发明化合物在大鼠中药物代谢动力学实验
[0276]
一、实验目的
[0277]
用sd大鼠,以液相色谱
‑
质谱(ls/ms/ms)法测定了大鼠灌胃给予实施例2、实施例6、实施例25化合物后不同时刻血浆中药物浓度并计算相关药代参数,评价本发明实施例化合物在大鼠体内药代动力学特性。
[0278]
二、给药
[0279]
具体给药按照试验方案进行,详情见表3。
[0280]
表3.大鼠药代动力试验方案
[0281][0282]
三、采血时间及样品处理
[0283]
给药前及给药后10分钟、20分钟、0.5小时、1小时、2小时、4小时、8小时、12小时、24小时通过眼眶采血,每次采血0.2毫升,置于乙二胺四乙酸二钾(edta
‑
2k)试管中,12000转每分钟(rpm),离心5分钟,收集血浆,保存于
‑
20℃冰箱中备用。
[0284]
四、样品测试及数据分析
[0285]
采用液相色谱
‑
质谱(lc/ms/ms)方法测定小鼠血浆中化合物的含量。利用winnonlin5.3软件的非房室模型计算给药后化合物的药代动力学参数。
[0286]
五、试验结果
[0287]
实施例2、实施例6、实施例25化合物均有较好的代谢特征及较好的生物利用度。
[0288]
实验六、本发明实施例化合物的急毒实验
[0289]
一、实验目的和方法
[0290]
本实验目的是为了测试化合物在小鼠上的毒性效果。
[0291]
小鼠单次给予不同剂量的化合物11,观察14天,记录动物死亡情况,中毒反应,体重变化,饮食,外观,行为等。终点解剖动物,取脏器,进行组织病理学检查。
[0292]
二、实验结果与结论
[0293]
本发明化合物的半数致死剂量(ld50)>1000毫克每公斤(mg/kg),安全性好。与对照组小鼠比较,给药组小鼠自给药日起14天内未见体重及行为异常,本发明化合物并未显示出明显毒性。
[0294]
实验七、本发明实施例5、实施例15、实施例26化合物在人弥散性巨型b细胞淋巴瘤细胞(wsu
‑
dlcl2细胞)小鼠移植瘤模型中的药效实验
[0295]
一、实验步骤
[0296]
无菌条件下,将对数生长期的弥散性巨型b细胞淋巴瘤细胞(wsu
‑
dlcl2细胞系)消化并与基质胶(matrigel)混合后移植于联合重度免疫缺陷(cb17/scid)小鼠背部右侧皮下,每只小鼠接种1*107个细胞,体积100微升(μl),接种后,将小鼠根据肿瘤大小均衡随机分成5组,每组6只进行体内药效实验,阳性对照组为epz
‑
6438,阴性对照组给等量的溶媒。
具体设计见表4。
[0297]
表4.化合物体内药效实验
[0298][0299][0300]
二、实验结果及结论
[0301]
epz
‑
6438在150毫克每千克(mg/kg)的给药浓度下对肿瘤生长抑制结果只有52%,实施例5、实施例15、实施例26化合物在150毫克每千克(mg/kg)的给药浓度下对肿瘤生长抑制率分别达到56%、63%、65%;表明本发明实施例5、15、26化合物在弥散性巨型b细胞淋巴瘤细胞(wsu
‑
dlcl2细胞系)异种移植模型中比epz
‑
6438有更强的肿瘤生长抑制作用。
[0302]
实验八、本发明实施例20化合物在人弥散性巨型b细胞淋巴瘤细胞(wsu
‑
dlcl2细胞)小鼠移植瘤模型中的药效实验
[0303]
一、实验步骤
[0304]
无菌条件下,将对数生长期的人弥散性巨型b细胞淋巴瘤细胞(wsu
‑
dlcl2细胞)消化并与基质胶(matrigel)混合后移植于联合重度免疫缺陷(cb17/scid)小鼠背部右侧皮下,每只小鼠接种1*107个细胞,体积100微升(μl),接种后,将小鼠根据肿瘤大小均衡随机随机分成5组,每组6只进行体内药效实验,阳性对照组为epz
‑
6438,阴性对照组给等量的溶媒。具体设计见表5。
[0305]
表5.化合物体内药效实验
[0306][0307]
二、实验结果及结论
[0308]
epz
‑
6438在125mg/kg和250mg/kg的给药浓度下对肿瘤生长抑制率分别为47%、72%,实施例20化合物125mg/kg、250mg/kg、500mg/kg的给药浓度下对肿瘤生长抑制率分别为56%、85%、91%;表明本发明实施例20化合物在人弥散性巨型b细胞淋巴瘤细胞(wsu
‑
dlcl2细胞系)异种移植模型中比epz
‑
6438更有更强的肿瘤生长抑制作用。
[0309]
实验九、本发明实施例23化合物在人弥漫性大细胞淋巴瘤b淋巴细胞(pfeiffer细胞)小鼠移植瘤模型中的药效实验
[0310]
一、实验步骤
[0311]
无菌条件下,将对数生长期的人弥漫性大细胞淋巴瘤b淋巴细胞(pfeiffer细胞)消化并与基质胶(matrigel)混合后移植于联合重度免疫缺陷(beige scid)小鼠背部右侧皮下,每只小鼠接种1*107个细胞,体积100微升(μl),接种后,将小鼠根据肿瘤大小均衡随机分成5组,每组6只进行体内药效实验,阳性对照组为epz
‑
6438,阴性对照组给等量的溶媒。具体设计见表6。
[0312]
表6.化合物体内药效实验
[0313][0314]
二、实验结果及结论
[0315]
epz
‑
6438在50mg/kg和100mg/kg的给药浓度下对肿瘤生长抑制率分别为53%、81%,实施例23化合物50mg/kg、100mg/kg的给药浓度下对肿瘤生长抑制率分别为78%、92%;表明本发明实施例23化合物在人弥漫性大细胞淋巴瘤b淋巴细胞(pfeiffer细胞)异种移植模型中比epz
‑
6438更有更强的肿瘤生长抑制作用。
[0316]
实验十、本发明实施例18化合物在人肾癌细胞系(g401细胞)小鼠移植瘤模型中的药效实验
[0317]
一、实验步骤
[0318]
无菌条件下,将对数生长期的人肾癌细胞(g401细胞)消化并与基质胶(matrigel)混合后移植于联合重度免疫缺陷(scid)小鼠背部右侧皮下,每只小鼠接种1*107个细胞,体积100微升(μl),接种后,将小鼠根据肿瘤大小均衡随机分成5组,每组6只进行体内药效实验,阳性对照组为epz
‑
6438,阴性对照组给等量的溶媒。具体设计见表7。
[0319]
表7.化合物体内药效实验
[0320][0321]
[0322]
二、实验结果及结论
[0323]
epz
‑
6438在125mg/kg和250mg/kg的给药浓度下对肿瘤生长抑制率分别为41%、66%,实施例18化合物125mg/kg和250mg/kg的给药浓度下对肿瘤生长抑制率分别为50%、71%;表明本发明实施例18化合物在人肾癌细胞系(g401细胞)异种移植模型中比epz
‑
6438更有更强的肿瘤生长抑制作用。
[0324]
实验十一、本发明实施例18化合物在人胃癌细胞(nci
‑
n87细胞)小鼠移植瘤模型中的药效实验
[0325]
一、实验步骤
[0326]
无菌条件下,将对数生长期的人胃癌细胞(nci
‑
n87细胞)消化并与基质胶(matrigel)混合后移植于联合重度免疫缺陷(scid)小鼠背部右侧皮下,每只小鼠接种1*107个细胞,体积100微升(μl),接种后,将小鼠根据肿瘤大小均衡随机分成5组,每组6只进行体内药效实验,阳性对照组为epz
‑
6438,阴性对照组给等量的溶媒。具体设计见表8。
[0327]
表8.化合物体内药效实验
[0328][0329]
二、实验结果及结论
[0330]
epz
‑
6438在125mg/kg和250mg/kg的给药浓度下对肿瘤生长抑制率分别为42%、63%,实施例18化合物125mg/kg和250mg/kg的给药浓度下对肿瘤生长抑制率分别为51%、68%;表明本发明实施例18化合物在人胃癌细胞(nci
‑
n87细胞)异种移植模型中比epz
‑
6438更有更强的肿瘤生长抑制作用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。