1.本发明涉及制药领域,特别是涉及一种美洛昔康的注射剂及其制备方法和用途。
背景技术:
2.术后疼痛(postoperative pain)是手术后发生的急性疼痛。积极采取有效的术后镇痛措施缓解疼痛,是加速康复、提高患者的舒适度和生活质量的关键环节。(临床药师术后疼痛管理指引,广东药学会,2019.1)目前,阿片类药物是临床最为常用的一类治疗中、重度疼痛的药物,通过与外周及中枢神经系统(脊髓及脑)阿片受体结合发挥镇痛作用。强阿片药包括吗啡、芬太尼、哌替啶、舒芬太尼等,具有镇痛作用强,无器官毒性和封顶效应的优点。但阿片类药物可能导致呼吸抑制、恶心、呕吐、肠梗阻等。同时,阿片类药物具有潜在的成瘾性和依赖性。为此,非阿片类药物一直是术后镇痛,特别是术后急性疼痛控制研究的热点。
3.美洛昔康(meloxicam,mlx)是一种新型的非甾体抗炎药(nsaids),1996年由德国勃林格殷格翰公司在南非上市。作为环氧合酶
‑
2(cox
‑
2)选择性抑制剂,美洛昔康具有优异的抗炎和镇痛效果,广泛用于骨关节炎、类风湿关节炎、强直性脊柱炎以及其他肌肉或骨骼疼痛(例如腰痛等)的治疗,也可用于患者术后轻、中度疼痛的镇痛。由于美洛昔康对cox
‑
2的抑制活性大于对于环氧合酶
‑
1(cox
‑
1)的抑制活性。因此,美洛昔康的血液(血小板)、消化道、肾脏和心血管等的副作用比cox
‑
1抑制剂小。与萘普生、布洛芬、吡罗昔康等传统的nsaids相比,美洛昔康的抗炎镇痛效果相似,而胃肠道耐受性显著提高。患者用药期间几乎没有胃肠穿孔、出血、溃疡情况,恶心、呕吐现象也较少。因此,美洛昔康有望成为一种非阿片类的镇痛药物,用于术后中、重度疼痛控制,但高脂溶性(logp=2.47)和极低的水溶性,导致美洛昔康口服后达峰时间较长,约需4~5小时才能达到血药浓度的峰值,限制其在急性中重度疼痛中的应用。
4.为了加快美洛昔康的起效速度,研究人员开发了多种不同的制剂。在全身镇痛方面,目前仅有一个美洛昔康纳米晶注射剂(商品名为anjeso)上市。每日静脉注射一次,用于术后镇痛。该纳米晶体注射液经快速静脉推注后,迅速起效,显著缓解中度到重度疼痛。但其静脉快速推注、纳米晶的物理不稳定性均使其具有潜在威胁。在局部镇痛制剂方面,主要包括美洛昔康透皮给药制剂,如凝胶、脂质体、贴片和微乳液等。透皮给药制剂可在给药局部维持一定的药物浓度并保持较低的血浆浓度,避免对胃肠道的刺激,减少全身不良反应。但是透皮给药难以克服皮肤角质层屏障,美洛昔康的高脂溶性也影响了到达真皮层的药物的量。截止2016年,成功上市的美洛昔康透皮吸收剂只有一个。(jianmin chen,yunhua gao.strategies for meloxicam delivery to and across the skin:a review[j].drug delivery,2016,23(8).)。
技术实现要素:
[0005]
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种美洛昔康的注射剂及
其制备方法和用途,用于解决现有技术中的问题。
[0006]
为实现上述目的及其他相关目的,本发明是通过以下技术方案获得的。
[0007]
本发明提供一种用于装载美洛昔康的脂质体,所述脂质体内含有内水相,所述内水相为含有金属离子的盐溶液,所述美洛昔康能够与所述金属离子络合。
[0008]
优选地,所述金属离子选自钙离子、锌离子、铜离子、镁离子、锰离子或钴离子。
[0009]
优选地,所述盐溶液的ph值为6.0~8.0。
[0010]
优选地,所述脂质体包括磷脂和胆固醇中的一种或两种。
[0011]
本发明提供一种美洛昔康的注射剂,所述注射剂的原料组分包括:美洛昔康、外水相和如上述所述的脂质体;所述外水相至少含有水。
[0012]
优选地,以所述注射剂的体积为基准计,所述美洛昔康的浓度大于等于0.01mg/ml;和/或,外水相还含有葡甲胺、蔗糖或葡萄糖中的一种或多种。
[0013]
优选地,所述美洛昔康装载于所述脂质体内。
[0014]
本发明还公开了一种如上述所述的注射剂的制备方法,将美洛昔康、外水相与脂质体混合。
[0015]
本发明还公开了一种如上述所述的注射剂作为注射用镇痛剂的用途。
[0016]
本发明还公开了一种抗炎制剂,包括如上述所述的注射剂和细胞毒性药物,或如上述所述的注射剂和免疫药物。
[0017]
优选地,所述细胞毒性药物为阿霉素或盐酸阿霉素。
[0018]
优选地,所述细胞毒性药物和/或所述免疫药物装载在所述脂质体内。
[0019]
本发明还公开了一种如上述所述抗炎制剂用于制备治疗肿瘤的药物的用途。
[0020]
本技术中所述注射剂载药量高、储存稳定性好,静脉注射进入血液循环后,美洛昔康可快速释放,发挥全身镇痛作用;也可用于关节腔等局部注射,在较长时间内维持局部的药物浓度,改善局部镇痛和抗炎效果,减少全身给药的用量。
附图说明
[0021]
图1美洛昔康(mlx)水溶液的标准曲线。
[0022]
图2美洛昔康和金属离子盐溶液混合后形成的沉淀。
[0023]
图3以醋酸钙为内水相的美洛昔康脂质体的冷冻透射电镜照片。
[0024]
图4以醋酸钙为内水相的美洛昔康脂质体的包封率、储存稳定性和释放曲线。
[0025]
图5以氯化钙为内水相的美洛昔康脂质体的包封率、储存稳定性和释放曲线。
[0026]
图6以醋酸锌为内水相的美洛昔康脂质体的包封率、储存稳定性和释放曲线。
[0027]
图7以硫酸铜为内水相的美洛昔康脂质体的包封率、储存稳定性和释放曲线。
[0028]
图8以氯化镁为内水相的美洛昔康脂质体的包封率、储存稳定性和释放曲线。
[0029]
图9以氯化钴为内水相的美洛昔康脂质体的包封率、储存稳定性和释放曲线。
[0030]
图10以氯化锰为内水相的美洛昔康脂质体的包封率、储存稳定性和释放曲线。
[0031]
图11以氯化锰为内水相的美洛昔康、阿霉素单载脂质体的包封率。
[0032]
图12以氯化锰为内水相的美洛昔康
‑
阿霉素共载脂质体的包封率。
[0033]
图13美洛昔康
‑
阿霉素共载脂质体对(耐阿霉素)k562细胞的抑制作用。
[0034]
图14模拟局部注射给药不同内水相脂质体累计释放速率。
具体实施方式
[0035]
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。
[0036]
在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。下列实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。
[0037]
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
[0038]
本发明整个发明构思在于提供了一种可用于注射的美洛昔康脂质体制剂。不同于现有的通过薄膜分散法制备的美洛昔康脂质体,本法提供的美洛昔康该脂质体制剂采用主动载药的方法,利用美洛昔康与脂质体的内水相中金属离子结合形成不溶性盐(沉淀),使美洛昔康可以有效地进入脂质体内水相,达到稳定装载的目的。
[0039]
本技术中首先提供了一种用于装载美洛昔康的脂质体,所述脂质体内含有内水相,所述内水相为含有金属离子的盐溶液,所述美洛昔康能够与所述金属离子络合形成不溶性盐。优选地,所述金属离子选自钙离子、锌离子、铜离子、镁离子、锰离子或钴离子。优选地,所述盐溶液的ph值为6.0~8.0。优选地,所述脂质体包括磷脂和胆固醇中的一种或两种。本技术中申请人意外发现该脂质体具有较高的美洛昔康载药量和良好的储存稳定性。
[0040]
本技术中还公开了一种美洛昔康的注射剂,所述注射剂的原料组分包括:美洛昔康、外水相和如上述所述的脂质体;所述外水相至少含有水。优选地,以所述注射剂的体积为基准计,所述美洛昔康的浓度大于等于0.01mg/ml。优选地,外水相还含有葡甲胺、蔗糖或葡萄糖中的一种或多种。
[0041]
优选地,所述美洛昔康装载于所述脂质体内。
[0042]
上述这种注射剂的制备方法为:将美洛昔康、外水相与脂质体混合。
[0043]
在大体积稀释(例如静脉注射)时,美洛昔康可以快速释放,用于急性疼痛的控制以及中重度疼痛的全身镇痛;通过局部注射时,例如注射到关节腔内,美洛昔康脂质体可以缓慢释放,在较长时间内维持局部的药物浓度,改善局部镇痛和抗炎效果,并减少全身给药的用量。
[0044]
此外,新的研究表明,肿瘤微环境中的炎症因子如肿瘤坏死因子(tnf
‑
α)和白介素
‑
6(il
‑
6),通过复杂的信号通路,影响了t细胞等免疫细胞在肿瘤中的渗透(infiltration)(jose r.conejo
‑
garcia,breaking barriers for t cells by targeting the epha2/tgf
‑
b/cox
‑
2axis in pancreatic cancer,j clin invest.2019;129(9):3521
‑
3523),或者影响了肿瘤细胞对于细胞毒性药物的耐药性等,从而降低肿瘤免疫治疗或化疗的效果。考虑到美洛昔康的另一个主要作用是通过抑制cox
‑
2起到抗炎的效果,因此,本发明提出的这种可稳定载药的美洛昔康脂质体有望通过与细胞毒性药物或免
疫药物的联合使用,通过纠正肿瘤炎性微环境,提高免疫治疗或化疗的效果。
[0045]
在一个具体的实施方式中,所述细胞毒性药物为阿霉素或者盐酸阿霉素。
[0046]
在一个优选的实施方式中,所述细胞毒性药物和/或所述免疫药物装载在所述脂质体内。
[0047]
本技术中还公开了上述所述脂质体的具体的制备方法可以参照现有技术中常规的包覆法来制备。具体地,可以采用如下所述的方法:将脂质的有机溶剂溶液与内水相混合得到脂质体混悬液;然后通过透析除去脂质体外水相中的金属盐。一般的外水相的ph为6~7。由此,脂质体内外水相之间具有一种的ph梯度和金属盐浓度梯度。在一个优选的实施方式中,所述制备脂质体混悬液的温度为45~85℃。
[0048]
实施例1
[0049]
本实施例为美洛昔康的检测方法。
[0050]
本实施例中建立美洛昔康的标准曲线,具体如下。
[0051]
采用紫外(ultraviolet,uv)检测分析方法:检测仪器为tecan200pro;检测波长为362nm;检测温度为25℃;检测孔板为96well plates,uv
‑
transparent;检测体积为200μl。
[0052]
(1)精密称量2mg美洛昔康,加入到2ml氢氧化钠溶液(ph~10)中,涡旋至完全溶解,制备成1mg/ml的美洛昔康储备液。
[0053]
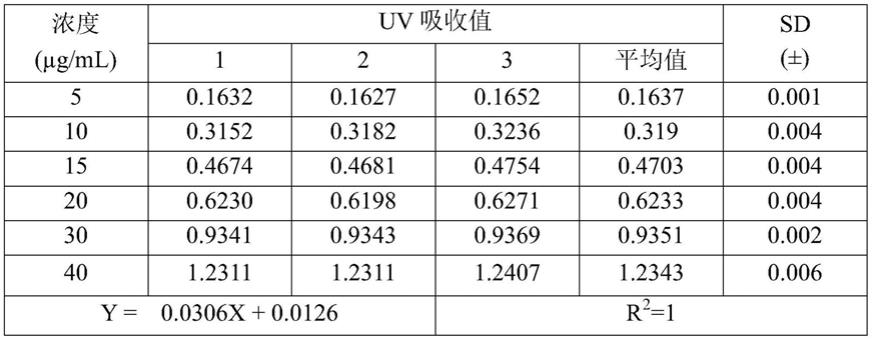
(2)将美洛昔康水溶液与超纯水混合,梯度稀释,得到浓度为5μl/ml、10μl/ml、15μl/ml、20μl/ml、30μl/ml和40μl/ml的美洛昔康标准溶液。采用uv法检测上述浓度的美洛昔康标准溶液,检测结果如表1所示。
[0054]
表1不同浓度的美洛昔康水溶液uv检测值
[0055][0056]
根据表1所示的结果建立标准曲线。工作曲线如图1所示。美洛昔康水溶液的uv标准曲线为y=0.0306x 0.0126(n=6),r2=1,拟合良好。
[0057]
实施例2
[0058]
本实施例为美洛昔康的增溶以及美洛昔康与金属离子形成不溶性盐。
[0059]
2.1羟丙基
‑
β
‑
环糊精(hpcd)对美洛昔康的增溶作用
[0060]
将一定量hpcd(上海士锋生物科技有限公司)用去离子水溶解,配制成浓度分别为0%、10%、20%、30%的hpcd溶液。取2ml不同浓度的hpcd溶液,分别加入美洛昔康约2mg,涡旋5min后在37℃恒温混匀仪(thermomixer c,eppendorf)上震摇24h。取出后离心,测定上
清溶液中美洛昔康的紫外吸收值,按照实施例1中的工作曲线,计算美洛昔康在不同浓度的hpcd溶液中的溶解度。
[0061]
2.2葡甲胺对美洛昔康的增溶作用
[0062]
本实施例中使用的葡甲胺购自百灵威科技有限公司
[0063]
称取适量的葡甲胺和过量美洛昔康制备成含有葡甲胺10mg/ml、20mg/ml、30mg/ml的溶液,在37℃下,以500rpm震摇24h。取出后离心,测定上清溶液中美洛昔康的紫外吸收值,按照实施例1中的工作曲线,计算美洛昔康在不同浓度葡甲胺溶液中的溶解度。
[0064]
表2美洛昔康在不同增溶剂溶液中的饱和溶解度
[0065][0066]
如表2所示,hpcd对美洛昔康具有一定的增溶作用,且hpcd浓度越高,增溶作用越强。而葡甲胺对美洛昔康的增溶作用远大于hpcd。
[0067]
2.3美洛昔康溶液与金属离子结合,形成不溶性沉淀
[0068]
按照2.2中的方法,制备葡甲胺增溶的美洛昔康溶液(浓度约为16mg/ml);配制浓度均为300mm的醋酸钙、氯化钙、醋酸锌、硫酸铜、氯化镁、氯化钴和氯化锰溶液。将美洛昔康溶液(50μl)分别加入到各金属离子溶液(1ml)中。美洛昔康均能够与这些金属离子结合,形成不溶性沉淀(图2)
[0069]
实施例3
[0070]
本实施例为以醋酸钙溶液为内水相的美洛昔康脂质体的制备和表征。
[0071]
本实施例中所使用的磷脂均购自德国lipoid公司。具体为氢化豆磷脂(hspc,分子量783.8);聚乙二醇化磷脂具体为二硬脂酰磷脂酰乙醇胺
‑
聚乙二醇2000(dspe
‑
peg2000);胆固醇(chol,分子量386.7);使用的美洛昔康购自梯希爱(上海)化成工业发展有限公司。
[0072]
需要说明的是,在后续的实施例中所使用的磷脂均为lipoid公司的氢化豆磷脂;所使用的聚乙二醇化磷脂均为lipoid公司的二硬脂酰磷脂酰乙醇胺
‑
聚乙二醇;所使用的胆固醇均为lipoid的胆固醇;所使用的美洛昔康均购自梯希爱(上海)化成工业发展有限公司。
[0073]
3.1以醋酸钙溶液为内水相的美洛昔康脂质体的制备
[0074]
制备方法如下:
[0075]
(1)分别配置浓度为150mm、300mm、500mm的醋酸钙水溶液,用盐酸调节ph为8.0;
[0076]
(2)将氢化豆磷脂:二硬脂酰磷脂酰乙醇胺
‑
聚乙二醇2000:胆固醇按照3:1:1的摩尔比在无水乙醇中熔融,得到脂质的乙醇混合溶液;
[0077]
(3)在步骤(2)所得脂质的乙醇混合溶液中加入3ml步骤(1)中制备的醋酸钙溶液,并置于60℃中搅拌水浴30分钟,充分水合脂质,得到较均匀的脂质体混悬液;
[0078]
(4)利用脂质体挤出仪,将步骤(3)所得脂质体混悬液依次挤压通过特定孔径的碳膜,进而控制空白脂质体的粒径大小与均匀性;
[0079]
(5)将步骤(4)所制得的脂质体置于截留分子量为10kda的透析袋中,以与人体等渗的10%的蔗糖水溶液作为透析介质,样品与透析介质体积比为1:1000,透析,除去脂质体
外水相中的醋酸钙,得到由10%的蔗糖组成的外水相,以及由醋酸钙溶液组成的内水相,和磷脂双分子层构成空白脂质体,脂质体内水相具有一定的ph和醋酸钙浓度梯度。具体地,脂质双分子层内水相为醋酸钙水溶液、脂质双分子层外水相为蔗糖水溶液(ph为6~7)。
[0080]
(6)按照实例2制备的美洛昔康
‑
葡甲胺溶液(摩尔比为1:1)的方法,制备成含美洛昔康8mg/ml的葡甲胺水溶液。
[0081]
(7)按照药脂比0.05、0.1、0.2,取适量的美洛昔康溶液与空白脂质体混合,60℃孵育30min,制成美洛昔康脂质体溶液。
[0082]
3.2.以醋酸钙溶液为内水相的美洛昔康脂质体的表征
[0083]
3.2.1以醋酸钙溶液为内水相的美洛昔康脂质体的粒径
[0084]
将制备得到的美洛昔康脂质体,用去离子水稀释100倍,利用zetasizer zs90(马尔文公司,英国)测定脂质体的粒径。如表3所示,美洛昔康脂质体粒径均在100nm左右,粒度分布(pdi)均小于0.1。
[0085]
3.2.2以醋酸钙溶液为内水相的美洛昔康脂质体的形貌
[0086]
取以300mm醋酸钙溶液为内水相制备的美洛昔康脂质体(药脂比0.1)约5μl,滴加到300目lacy铜网(tedpella,美国)。吸去过量溶液,快速放入液态乙烷中。采用冷冻透射电镜(feitalos,美国thermo公司),在120kv电压下,观察脂质体形貌。结果如图3(b为空白脂质体、c为载药脂质体)所示。美洛昔康脂质体为球形,平均粒径在100nm左右;在一些脂质体内壁有“碗装”结构出现(图3中白色箭头所示),可能是美洛昔康作为一种难溶性化合物,进入脂质体内水相后,沉淀在脂质膜内侧。因此可获得较高的包封率和载药量,并且不易泄露,具有较好的储存稳定性。
[0087]
3.2.2以醋酸钙溶液为内水相的美洛昔康脂质体的包封率
[0088]
将制备得到的美洛昔康脂质体用超纯水稀释50倍,加入适量dowex树脂(西格玛奥德里奇(sigma
‑
aldrich)公司),充分振摇,吸附未包裹的美洛昔康。静置后取上清200μl,采用实施例1中的紫外检测方法,测定加入树脂前后脂质体中的美洛昔康的吸光度值,并计算含量。
[0089]
美洛昔康脂质体的包封率(ee)按以下公式计算:
[0090][0091]
其中,minner是树脂吸附游离药物后的脂质体制剂中美洛昔康的量,即被脂质体包封的美洛昔康的量;mtotal是树脂吸附前美洛昔康脂质体制剂中美洛昔康的总量。结果如表3所示,该处方美洛昔康脂质体具有较高包封率。其中,内水相中醋酸钙浓度从150mm增加到300mm时,脂质体的载药能力随醋酸钙浓度的提高而显著增加,但是超过300mm后,包封率增加趋缓。
[0092]
表3以醋酸钙溶液为内水相的美洛昔康脂质体粒径和包封率
[0093][0094]
3.3以醋酸钙溶液为内水相的美洛昔康脂质体储存稳定性
[0095]
取上述内水相为醋酸钙溶液的美洛昔康脂质体,置于4℃冰箱保存,分别在一定天数后,取样,稀释50倍,按照3.2.2中方法测包封率,考察以醋酸钙为内水相的美洛昔康脂质体稳定性。如图4(b)所示,三种不同浓度的醋酸钙为内水相处方在30天内均无明显泄露,故具有较好的储存稳定性。
[0096]
3.4以醋酸钙溶液为内水相的美洛昔康脂质体体外释放
[0097]
取上述内水相为不同浓度醋酸钙溶液的美洛昔康脂质体,以生理盐水为释放介质,加入足量的dowex树脂吸附游离药物,形成漏槽条件,于37℃100rpm摇床(thz
‑
c恒温振荡器,中国)中震荡。在不同的时间点(0小时、0.5小时、1小时、2小时、3小时、4小时、5小时、6小时和24小时)取样,静置后取上清,uv法测定包裹在脂质体中的美洛昔康的含量,计算不同时间点美洛昔康的累积释放速率。如图4(c)所示,在4小时内,三种不同浓度的醋酸钙溶液为内水相的处方累计释药量均大于80%,在4小时后释放减慢。
[0098]
实施例4
[0099]
本实施例是以氯化钙溶液为内水相的美洛昔康脂质体的制备和表征。
[0100]
4.1以氯化钙溶液为内水相的美洛昔康脂质体的制备
[0101]
配制浓度分别为150mm、300mm、500mm的氯化钙水溶液,用盐酸调节ph为7.5;按照实施例3中3.1的制备方法,制备以氯化钙为内水相的美洛昔康脂质体。
[0102]
4.2以氯化钙溶液为内水相的美洛昔康脂质体的表征
[0103]
按照实施例3中3.2的方法,测定以氯化钙为内水相制备的美洛昔康脂质体的粒径和包封率。结果如表4所示,制备得到的脂质体粒径均在100
±
10nm。粒度分布(pdi)均小于0.1。氯化钙浓度从150mm增加到300mm时,脂质体的载药能力随氯化钙浓度的提高而显著增加,但是超过300mm后,包封率增加趋缓。
[0104]
表4以氯化钙溶液为内水相的美洛昔康脂质体粒径和包封率
[0105][0106]
4.3以氯化钙溶液为内水相的美洛昔康脂质体的储存稳定性
[0107]
按照实施例3中3.3的方法,取上述内水相中醋酸钠浓度分别为150mm、300mm和500mm的美洛昔康脂质体,置于4℃冰箱保存,考察以氯化钙为内水相的美洛昔康脂质体稳定性。如图5(b)所示,在4℃储存条件下,三种不同浓度氯化钙为内水相处方在30天内均无明显泄露,故具有较好的储存稳定性。
[0108]
4.4以氯化钙溶液为内水相的美洛昔康脂质体的体外释放速率
[0109]
取上述内水相为不同浓度氯化钙溶液的美洛昔康脂质体,按照实施例3中3.4的体外释药速率表征方法,计算不同时间点脂质体中美洛昔康的累积释药百分比。结果如图5(c)所示,在4小时内,三种不同浓度的氯化钙钙溶液为内水相的处方累计释药量均大于80%,在4小时后释放减慢。
[0110]
实施例5
[0111]
本实施例中以醋酸锌溶液为内水相的美洛昔康脂质体的制备和表征。
[0112]
5.1以醋酸锌溶液为内水相的美洛昔康脂质体的制备
[0113]
配制浓度分别为150mm、300mm、500mm的醋酸锌水溶液,用盐酸调节ph为6.0;按照实施例3中3.1的制备方法,制备以醋酸锌为内水相的美洛昔康脂质体。
[0114]
5.2以醋酸锌溶液为内水相的美洛昔康脂质体的表征
[0115]
按照实施例3中3.2的方法,测定以醋酸锌为内水相制备的美洛昔康脂质体的粒径和包封率。结果如表5所示,美洛昔康脂质体粒径均在155
±
5nm左右。粒度分布(pdi)均小于0.1。醋酸锌浓度从150mm增加到300mm时,脂质体的载药能力随醋酸锌浓度的提高而显著增加,但是超过300mm后,包封率增加趋缓。
[0116]
表5以醋酸锌溶液为内水相的美洛昔康脂质体粒径和包封率
[0117][0118]
5.3以醋酸锌溶液为内水相的美洛昔康脂质体的储存稳定性
[0119]
按照实施例3中3.3的方法,取上述内水相为不同浓度醋酸锌溶液的美洛昔康脂质体,置于4℃冰箱保存,考察以醋酸锌为内水相的美洛昔康脂质体稳定性,结果如图6(b)所示,三种不同浓度的醋酸锌为内水相的处方在30天未发生泄露,故具有良好的储存稳定性。
[0120]
5.4以醋酸锌溶液为内水相的美洛昔康脂质体的体外释放速率
[0121]
取上述内水相为不同浓度醋酸锌溶液的美洛昔康脂质体,按照实施例3中3.4的体外释药速率表征方法,计算不同时间点脂质体中美洛昔康的累积释药百分比。结果如图6(c)所示,在4小时内,三种不同浓度醋酸锌为内水相的处方累计释药量均大于70%,在4小时后释放减慢。
[0122]
实施例6
[0123]
实施例6以硫酸铜溶液为内水相的美洛昔康脂质体的制备和表征。
[0124]
6.1以硫酸铜溶液为内水相的美洛昔康脂质体的制备
[0125]
配制浓度分别为150mm、300mm、500mm的硫酸铜水溶液,用盐酸调节ph为7.0;按照实施例3中3.1的制备方法,制备以硫酸铜为内水相的美洛昔康脂质体。
[0126]
6.2以硫酸铜溶液为内水相的美洛昔康脂质体的表征
[0127]
按照实施例3中3.2的方法,测定以硫酸铜为内水相制备的美洛昔康脂质体的粒径和包封率。结果如表6所示,美洛昔康脂质体粒径均在130
±
5nm左右。粒度分布(pdi)均小于0.1。硫酸铜浓度从150mm增加到300mm时,脂质体的载药能力随硫酸铜浓度的提高而显著增加,但是超过300mm后,包封率增加趋缓。
[0128]
表6以硫酸铜溶液为内水相的美洛昔康脂质体粒径和包封率
[0129][0130]
6.3以硫酸铜溶液为内水相的美洛昔康脂质体的储存稳定性
[0131]
按照实施例3中3.3的方法,取上述内水相为不同浓度硫酸铜溶液的美洛昔康脂质体,置于4℃冰箱保存,考察以硫酸铜为内水相的美洛昔康脂质体稳定性,结果如图7(b)所示,三种不同浓度的硫酸铜为内水相的处方在30天未发生泄露,故具有良好的储存稳定性。
[0132]
6.4以硫酸铜溶液为内水相的美洛昔康脂质体的体外释放速率
[0133]
取上述内水相为不同浓度硫酸铜溶液的美洛昔康脂质体,按照实施例3中3.4的体外释药速率表征方法,计算不同时间点脂质体中美洛昔康的累积释药百分比。结果如图7(c)所示,在4小时内,三种不同浓度硫酸铜为内水相的处方累计释药量均大于70%,在4小时后释放减慢。
[0134]
实施例7
[0135]
本实施例为以氯化镁溶液为内水相的美洛昔康脂质体的制备和表征。
[0136]
7.1以氯化镁溶液为内水相的美洛昔康脂质体的制备
[0137]
配制浓度分别为150mm、300mm、500mm的氯化镁水溶液,用盐酸调节ph为6.5;按照实施例3中3.1的制备方法,制备以氯化镁为内水相的美洛昔康脂质体。
[0138]
7.2以氯化镁溶液为内水相的美洛昔康脂质体的表征
[0139]
按照实施例3中3.2的方法,测定以氯化镁为内水相制备的美洛昔康脂质体的粒径和包封率。结果如表7所示,美洛昔康脂质体粒径均在110
±
5nm左右。粒度分布(pdi)均小于0.1。氯化镁浓度从150mm增加到300mm时,脂质体的载药能力随氯化镁浓度的提高而显著增加,但是超过300mm后,包封率增加趋缓。
[0140]
表7以氯化镁溶液为内水相的美洛昔康脂质体粒径和包封率
[0141][0142]
7.3以氯化镁溶液为内水相的美洛昔康脂质体的储存稳定性
[0143]
按照实施例3中3.3的方法,取上述内水相为不同浓度氯化镁溶液的美洛昔康脂质体,置于4℃冰箱保存,考察以氯化镁为内水相的美洛昔康脂质体稳定性,结果如图8(b)所示,三种不同浓度的氯化镁为内水相的处方在30天未发生明显泄露,故具有良好的储存稳定性。
[0144]
7.4以氯化镁溶液为内水相的美洛昔康脂质体的体外释放速率
[0145]
取上述内水相为不同浓度氯化镁溶液的美洛昔康脂质体,按照实施例3中3.4的体外释药速率表征方法,计算不同时间点脂质体中美洛昔康的累积释药百分比。结果如图8(c)所示,在4小时内,三种不同浓度氯化镁为内水相的处方累计释药量均大于70%,在4小时后释放减慢。
[0146]
实施例8
[0147]
本实施例是以氯化钴溶液为内水相的美洛昔康脂质体的制备和表征。
[0148]
8.1以氯化钴溶液为内水相的美洛昔康脂质体的制备
[0149]
配制浓度分别为150mm、300mm、500mm的氯化钴水溶液,用盐酸调节ph为6.0;按照实施例3中3.1的制备方法,制备以氯化钴为内水相的美洛昔康脂质体。
[0150]
8.2以氯化钴溶液为内水相的美洛昔康脂质体的表征
[0151]
按照实施例3中3.2的方法,测定以氯化钴为内水相制备的美洛昔康脂质体的粒径和包封率。结果如表8所示,美洛昔康脂质体粒径均在120
±
5nm左右。粒度分布(pdi)均小于0.1。氯化钴浓度从150mm增加到300mm时,脂质体的载药能力随氯化钴浓度的提高而显著增加,但是超过300mm后,包封率增加趋缓。
[0152]
表8以氯化钴溶液为内水相的美洛昔康脂质体粒径和包封率
[0153][0154]
8.3以氯化钴溶液为内水相的美洛昔康脂质体的储存稳定性
[0155]
按照实施例3中3.3的方法,取上述内水相为不同浓度氯化钴溶液的美洛昔康脂质体,置于4℃冰箱保存,考察以氯化钴为内水相的美洛昔康脂质体稳定性,结果如图9(b)所示,三种不同浓度的氯化钴为内水相的处方在30天未发生明显泄露,故具有良好的储存稳定性。
[0156]
8.4以氯化钴溶液为内水相的美洛昔康脂质体的体外释放速率
[0157]
取上述内水相为不同浓度氯化钴溶液的美洛昔康脂质体,按照实施例3中3.4的体外释药速率表征方法,计算不同时间点脂质体中美洛昔康的累积释药百分比。结果如图9(c)所示,在4小时内,三种不同浓度氯化钴为内水相的处方累计释药量均大于70%,在4小时后释放减慢。
[0158]
实施例9
[0159]
本实施例为以氯化锰溶液为内水相的美洛昔康脂质体的制备和表征。
[0160]
9.1以氯化锰溶液为内水相的美洛昔康脂质体的制备
[0161]
配制浓度分别为150mm、300mm、500mm的氯化锰水溶液,用盐酸调节ph为6.5;按照实施例3中3.1的制备方法,制备以氯化锰为内水相的美洛昔康脂质体。
[0162]
9.2以氯化锰溶液为内水相的美洛昔康脂质体的表征
[0163]
按照实施例3中3.2的方法,测定以氯化锰为内水相制备的美洛昔康脂质体的粒径和包封率。结果如表9和图10所示,美洛昔康脂质体粒径均在120
±
5nm左右。粒度分布(pdi)均小于0.1。氯化锰浓度从150mm增加到300mm时,脂质体的载药能力随氯化锰浓度的提高而显著增加,但是超过300mm后,包封率增加趋缓。
[0164]
表9以氯化锰溶液为内水相的美洛昔康脂质体粒径和包封率
[0165][0166][0167]
9.3以氯化锰溶液为内水相的美洛昔康脂质体的储存稳定性
[0168]
按照实施例3中3.3的方法,取上述内水相为不同浓度氯化锰溶液的美洛昔康脂质体,置于4℃冰箱保存,考察以氯化锰为内水相的美洛昔康脂质体稳定性,结果如图10(b)所示,三种不同浓度的氯化锰为内水相的处方在30天未发生明显泄露,故具有良好的储存稳定性。
[0169]
9.4以氯化锰溶液为内水相的美洛昔康脂质体的体外释放速率
[0170]
取上述内水相为不同浓度氯化锰溶液的美洛昔康脂质体,按照实施例3中3.4的体外释药速率表征方法,计算不同时间点脂质体中美洛昔康的累积释药百分比。结果如图10(c)所示,在4小时内,三种不同浓度氯化锰为内水相的处方累计释药量均大于70%,在4小时后释放减慢。
[0171]
实施例10
[0172]
本实施例为美洛昔康脂质体静脉注射后的镇痛作用。
[0173]
采用小鼠扭体模型评价美洛昔康脂质体静脉注射后的镇痛效果。
[0174]
取50只spf级昆明小鼠,体重为18~22g。随机分为4组,每组10只,雌雄各半。分为阴性对照组(生理盐水)、阳性对照组(美洛昔康注射液,勃林格殷格翰,)、美洛昔康混悬液组、美洛昔康脂质体组和空白脂质体组。其中,脂质体组为实施例3中以醋酸钙为内水相制备的空白脂质体和美洛昔康载药脂质体(约2mg/ml)。除美洛昔康混悬液为腹腔给药外,其余各组均为尾静脉给药。美洛昔康给药剂量均为2mg/kg。
[0175]
各组注射药物20min后,腹腔注射1%冰醋酸,制作腹痛病理模型,并将小鼠置于倒置的量筒内,观察记录15min内的扭体次数,并计算抑制率:抑制率=(给药组平均扭体次数-阴性对照组平均扭体次数)/阴性对照组平均扭体次数
×
100%。
[0176]
采用单因素方差分析(one
‑
way anova)进行组间差异性分析。以p<0.05为差异有显著性,p<0.01为差异有极显著性。结果如表10所示。
[0177]
表10.对醋酸所致的小鼠扭体反应的抑制作用(美洛昔康剂量为2mg/kg)
[0178][0179][0180]
结果表明,与混悬液相比,美洛昔康脂质体静脉注射后可在血液中快速释放药物,表现出与注射液相似的镇痛效果。
[0181]
实施例11
[0182]
本实施例为以氯化锰为内水相的美洛昔康
‑
阿霉素共载药脂质体的制备和表征。
[0183]
11.1以氯化锰溶液为内水相的美洛昔康
‑
阿霉素共载药脂质体的制备
[0184]
(1)配制浓度为300mm的氯化锰水溶液,用盐酸调节ph为7;
[0185]
(2)按照实施例3中3.1(1)~(5)的方法,制备氯化锰空白脂质体;
[0186]
(3)按照实例2中的方法,制备美洛昔康
‑
葡甲胺水溶液(浓度为8mg/ml);
[0187]
(4)精密称取盐酸阿霉素10mg,10%蔗糖溶解至1ml,制成10mg/ml盐酸阿霉素溶液。
[0188]
(5)按照总药脂比0.1、0.2和0.3(美洛昔康与盐酸阿霉素摩尔比1:1),分别向空白脂质体中加入盐酸阿霉素溶液和美洛昔康溶液。60℃孵育30min,制成美洛昔康
‑
阿霉素共载药脂质体溶液。
[0189]
另外,采用(2)中的氯化锰脂质体,按照0.05、0.1和0.15的药脂比,分别向脂质体中加入美洛昔康溶液或盐酸阿霉素溶液,制备美洛昔康单载脂质体或者阿霉素单载脂质体。
[0190]
11.2以氯化锰溶液为内水相的美洛昔康
‑
阿霉素共载脂质体的表征
[0191]
按照实施例3中3.2的方法,测定以氯化锰为内水相制备的美洛昔康
‑
阿霉素共载脂质体的粒径和包封率(表11和图12),并与单载美洛昔康或阿霉素的脂质体的粒径和包封率(表12和图11)进行比较共载药脂质体粒径均在110
±
5nm左右。粒度分布(pdi)均小于0.1。阿霉素与美洛昔康共载,显著提高阿霉素的包封率。其中,图11中药脂比是指单个药物与脂质的摩尔比。图12中,药脂比是指美洛昔康与阿霉素总摩尔量与脂质摩尔量的比值。
[0192]
表11以氯化锰溶液为内水相的美洛昔康
‑
阿霉素共载脂质体粒径和包封率
[0193]
[0194]
表12.以氯化锰溶液为内水相,单载美洛昔康或单载阿霉素的脂质体的粒径和包封率
[0195][0196]
实施例12
[0197]
本实施例为美洛昔康
‑
阿霉素共载药脂质体对耐阿霉素k562细胞的细胞毒性实验:
[0198]
(1)k562细胞以每孔1
×
105个浓度接种于96孔板中,加入不同浓度的阿霉素单载脂质体、美洛昔康单载脂质体、美洛昔康
‑
阿霉素共载脂质体,孵育48h。孵育结束后,每孔加入10ul cck
‑
8试剂(碧云天),孵育4h。孵育结束后,在波长为450nm处测定各孔吸光度值。
[0199]
(2)耐阿霉素k562细胞以每孔1
×
105个浓度接种于96孔板中,加入不同浓度的阿霉素单载脂质体、美洛昔康单载脂质体以及美洛昔康
‑
阿霉素共载脂质体,孵育48h。孵育结束后,每孔加入10ul cck
‑
8试剂(碧云天),孵育4h。孵育结束后,在波长为450nm处测定各孔吸光度值。结果如图13,结果表明,美洛昔康与阿霉素共载脂质体对耐阿霉素k562细胞的抑制作用明显高于阿霉素单载脂质体,且美洛昔康单载脂质体对细胞无明显抑制作用。该结果提示,与美洛昔康共载有利于降低肿瘤细胞对阿霉素的耐药性。
[0200]
实施例13
[0201]
本实施例为考察脂质体局部注射后,在注射部位的药物释放速率。
[0202]
该实施例采用透析袋法测定脂质体药物的释放速率:将脂质体装入透析袋内,置于较小体积的释放介质中。由于只有从脂质体中释放出的药物才能从透析袋内扩散到释放介质中,因此,通过测定不同时间点释放介质中的药物浓度,即可获知脂质体的药物释放速率。需要注意的是,本实施例中的药物释放速率测定方法与实施例3.4中的方法不同。实施例3.4是将脂质体直接加入释放介质中,用以模拟脂质体静脉注射后,被血液大量稀释的情况;3.4中采用树脂吸附从脂质体中释放出的药物,以模拟药物从脂质体中释放后,立即被血液循环带走。而本实施例中采用透析袋法测定脂质体的药物释放速率,主要是为了模拟脂质体在注射局部较少的释放介质,以及释放出的药物会在局部停留一段时间的情况。
[0203]
具体步骤如下:
[0204]
取实施例3、实施例6、实施例7以及实施例9中内水相分别为300mm醋酸钙、硫酸铜、氯化镁、氯化锰的美洛昔康脂质体(药脂比均为0.1)各100μl,置于截止分子量为10000的透析袋(spectrumlabs,型号132570)中;将装有脂质体的透析袋浸泡于5ml释放介质(含有0.2%sds(w/v)的生理盐水)中,在37℃水浴中温和搅拌。分别在0小时、1小时、2小时、4小时、8小时、12小时、24小时、48小时、72小时、96小时取释放介质200μl,并补入相同体积的新
鲜释放介质。采用uv法测定溶液中美洛昔康的浓度(实施例1),计算不同时间点美洛昔康的累积释放速率。另取100μl浓度为100μg/ml的美洛昔康
‑
葡甲胺水溶液,加入透析袋中,相同方法测定释放速率。
[0205]
美洛昔康溶液和脂质体的累计释放速率如图14所示。在12小时内,美洛昔康脂质体的累计释药量均达到50%,之后药物释放速率平缓。而美洛昔康溶液则在约2hr即完全进入释放介质中,说明美洛昔康可以较快地扩散通过所使用的透析袋,透析袋不会阻滞美洛昔康的释放。
[0206]
美洛昔康脂质体累计释放速率按以下公式计算:
[0207][0208]
c
n
为第n各时间点所取样品浓度;v
n
为释放介质总体积;c
i
和v
i
分别为第i个时间点所取样品浓度以及取样体积;w为投药量;ee%为美洛昔康包封率。
[0209]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。