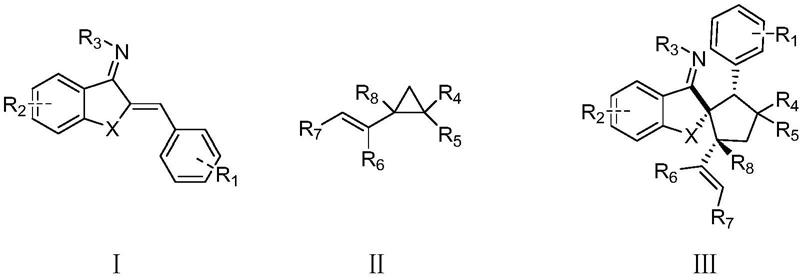
1.本发明涉及一种通过路易斯酸催化[3 2]环加成反应合成螺环类化合物的方法,属于有机合成技术领域。
背景技术:
[0002]
螺[苯并呋喃
‑
环戊烷]和螺[吲哚
‑
环戊烷]及其类似物结构广泛存在于天然产物及其衍生物中;同时螺环类化合物因具备刚性的空间结构,能够稳定分子的构型,和受体分子达到更好的结合,被广泛应用于药物设计中(luo,q.;wei,x.
‑
y.;yang,j.;luo,j.
‑
f.;liang,r.;tu,z.
‑
c.;cheng,y.
‑
x.j.nat.prod.2017,80,61
‑
70)。目前已经发展出了许多螺[苯并呋喃
‑
环戊烷]、螺[吲哚
‑
环戊烷]及其类似物的合成方法,如基于苯丙呋喃骨架的分子内反应、semipinacol重排等(liu,l.;lei,l.
‑
s.;zhan,z.
‑
s.;liu,s.
‑
z.;tu,y.
‑
q.;zhang,f.
‑
m.;zhang,x.
‑
m.;ma,a.
‑
j.;wang,s.
‑
h.chem.commun.2019,55,3789
‑
3792.),但这些方法存在前体制备复杂,产率偏低等问题。
[0003]
近年来,苯并呋喃骨架的氮杂二烯烃作为四元合成子被广泛应用于环合反应中,进行一系列[4 n](n≥2)环加成反应(trost,b.m.;zuo,z.
‑
j.angew.chem.,int.ed.2020,59,1243
‑
1247.)。在2020年,zhao课题组报道了一种通过苯并呋喃骨架的氮杂二烯烃和溴代丙二酸酯构建环丙烷的方法(fang,q.
‑
y.;yi,m.
‑
h.;wu,x.
‑
x.;z hao,l.
‑
m.org.lett.2020,22,5266
‑
5270.),实现了苯并呋喃骨架的氮杂二烯烃作为两元合成子的应用。
[0004]
乙烯基环丙烷因为具有较大的环张力而容易发生碳
‑
碳键的断裂,已经得到了化学家们深入、广泛的研究。shibata课题组开发了一种新的镁离子和碘负离子的复合物,可以活化乙烯基环丙烷。在镁离子的强路易斯酸催化作用下,碘负离子发生类共轭加成形成中间体,在没有受体的作用下中间体会发生分子内环合;在缺电子的异氰酸酯存在下,可以发生[3 2]环合反应。在他们的报道中,lii等不具备强路易斯酸性的试剂是没有催化效果的,必须在强路易斯酸的作用下才能进行[3 2]环合反应。这就使得催化剂路易斯酸的范围被限制,催化效率从而降低。
[0005]
因此以偕二酯基取代的乙烯基环丙烷和苯并呋喃或者吲哚骨架的氮杂二烯烃为底物,探究乙烯基环丙烷新的活化方法,从而构建出底物适用范围更广的[3 2]环加成反应合成螺[苯并呋喃
‑
环戊烷]和螺[吲哚
‑
环戊烷]化合物的合成方法,具有重要的意义。
技术实现要素:
[0006]
针对现有技术的不足,本发明提供了一种通过路易斯酸催化[3 2]环加成反应合成螺环类化合物的方法。本发明的方法以简单易得、价格低廉的路易斯酸作为催化剂,催化乙烯基环丙烷类化合物和苯并呋喃骨架或吲哚骨架的氮杂二烯的[3 2]环合反应,来制备螺[苯并呋喃
‑
环戊烷]和螺[吲哚
‑
环戊烷]类化合物,具有操作方便,底物适用范围广,原料
廉价易得等优点。
[0007]
本发明的技术方案如下:
[0008]
一种通过路易斯酸催化[3 2]环加成反应合成螺环类化合物的方法,包括步骤如下:
[0009]
于有机溶剂中,在路易斯酸催化下,氮杂二烯化合物ⅰ和乙烯基环丙烷化合物ⅱ发生[3 2]环加成反应,得到螺环类化合物ⅲ;
[0010][0011]
式ⅰ化合物结构式中,r1为氢、卤素、c1
‑
c3烷基、c1
‑
c3烷氧基、氰基或硝基;r2为氢、卤素、c1
‑
c3烷氧基;r3为对甲苯磺酰基或甲磺酰基;x为o或nac;
[0012]
式ⅱ化合物结构式中,r4与r5相同,为甲酸乙酯基、甲酸甲酯基中的一种,或r4、r5与其所连接的碳原子组成茚二酮基;r6为氢、苯基、甲基、乙基或异丙基;r7为氢、苯基、取代苯基、呋喃基、噻吩基、甲基、乙基、苄基或二甲基;所述取代苯基的取代基为c1
‑
c3烷氧基或卤素;r8为氢或c1
‑
c3烷基;
[0013]
式ⅲ化合物结构式中,取代基r1、r2、r3、x与式ⅰ化合物结构式中相同,取代基r4、r5、r6、r7、r8与式ⅱ化合物结构式中相同。
[0014]
根据本发明优选的,式ⅰ化合物结构式中,r1为氢、卤素、甲基、甲氧基、氰基或硝基;r2为氢、6
‑
氯基或6
‑
甲氧基;
[0015]
式ⅱ化合物结构式中,r7为取代苯基时,所述的取代苯基的取代基为4
‑
甲氧基或4
‑
溴基。
[0016]
根据本发明,所述式ⅱ化合物结构式中r7为二甲基时,式ⅱ化合物结构式如下式所示:
[0017][0018]
根据本发明优选的,所述有机溶剂为四氢呋喃、2
‑
甲基四氢呋喃、1,2
‑
二氯乙烷、乙酸乙酯、叔丁醇、乙腈或甲苯;所述有机溶剂的体积与氮杂二烯化合物ⅰ的摩尔数之比为5~20ml:1mmol。
[0019]
根据本发明优选的,所述路易斯酸为碘化镁、碘化镧、碘化锂、碘化钙或碘化铈;所述路易斯酸与氮杂二烯化合物ⅰ的摩尔比为0.01~0.5:1。
[0020]
根据本发明优选的,所述氮杂二烯化合物ⅰ和乙烯基环丙烷化合物ⅱ的摩尔比为1:1。
[0021]
根据本发明优选的,所述环加成反应的温度为50~70℃,进一步优选为60℃;所述
环加成反应时间为16
‑
24h。
[0022]
根据本发明优选的,所述环加成反应在惰性气体气氛下进行,所述的惰性气体为氮气或氩气。
[0023]
根据本发明,氮杂二烯化合物ⅰ和乙烯基环丙烷化合物ⅱ发生[3 2]环加成反应,可按常规分离纯化方法进行产物分离和表征。优选的,氮杂二烯化合物ⅰ和乙烯基环丙烷化合物ⅱ发生[3 2]环加成反应后所得反应液的后处理步骤如下:反应结束后,将反应液冷却至室温,然后用水淬灭反应,再用二氯甲烷萃取,将有机相用无水硫酸钠干燥,除去溶剂,得到的粗品再经柱色谱分离得到螺环类化合物ⅲ,洗脱剂为乙酸乙酯、二氯甲烷和石油醚的混合溶剂,其中乙酸乙酯、二氯甲烷和石油醚的混合溶剂中,乙酸乙酯、二氯甲烷和石油醚的体积比为1:0~3:5~20。
[0024]
根据本发明,所述苯并呋喃氮杂二烯烃化合物或吲哚类氮杂二烯烃化合物ⅰ由相应的苯并呋喃或吲哚不饱和酮及其类似物通过已知方法合成得到(参见文献:z.
‑
q.rong,m.wang,c.h.e.chow,y.zhao,chem.eur.j.2016,22,9483
–
9487.),反应路线如下所示:
[0025][0026]
其中,r1、r2、r3、x如上所述。
[0027]
根据本发明,所述乙烯基环丙烷化合物ⅱ由相应的丙二酸酯通过已知方法制备得到(参见文献:b.plietker,m.s.holzwarth,a.p.dieskau,j.am.chem.soc.2012,134,5048
‑
5051.,kimspielmanna,eleonoratosia,aur
é
lienlebrunb,gillesniela,arievan der leecrenata,marciade figueiredoa,jean
‑
marccampagnea,tetrahedron,2018,74(45),6497
‑
6511)
[0028]
r8为氢时,反应路线如下所示:
[0029][0030]
r8为c1
‑
c3烷基时,反应路线如下所示:
[0031][0032]
其中,r4、r5、r6、r7、r8如上所述。
[0033]
本发明的技术特点及有益效果如下:
[0034]
1、本发明以不同取代基取代的苯并呋喃骨架的氮杂二烯烃或吲哚骨架的氮杂二烯烃ⅰ和不同取代基取代的乙烯基环丙烷ⅱ作为原料,以简单易得、价格低廉的路易斯酸作为催化剂,通过[3 2]环加成反应制备得到螺[苯并呋喃
‑
环戊烷]和螺[吲哚
‑
环戊烷]类化合物ⅲ。本发明通过一种新的乙烯基环丙烷活化方法,从而使苯并呋喃和吲哚类氮杂二烯
烃类底物的范围扩大,生成更为复杂的螺[苯并呋喃
‑
环戊烷]和螺[吲哚
‑
环戊烷]类化合物。
[0035]
2、本发明的方法中所用的催化剂为简单易得、价格低廉的路易斯酸为催化剂,具有催化剂成本低,效率高等优点;本发明的通过路易斯酸催化的[3 2]环加成反应制备螺环类化合物的方法,具有操作方便,底物适用范围广泛,反应原料廉价易得等优点。
具体实施方式
[0036]
下面结合具体实施例对本发明作进一步的说明,但不限于此。
[0037]
同时下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂、材料和设备,如无特殊说明,均可从商业途径获得。
[0038]
实施例中所述收率为摩尔收率。
[0039]
实施例1
[0040]3‑
对甲苯磺酰亚胺基
‑
2'
‑
苯基
‑
5'
‑
(e)
‑
苯乙烯基
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
3',3'
‑
二甲酸甲酯(ⅲaa)的合成
[0041]
反应路线如下:
[0042][0043]
具体制备步骤如下:在手套箱中,氮气气氛下,向5ml圆底烧瓶中依次加入n
‑
((e)
‑2‑
((z)
‑
苯亚甲基)苯并呋喃
‑
3(2h)
‑
亚甲基)
‑4‑
甲基苯磺酰亚胺(ⅰa)(37.5mg,0.1mmol),2
‑
(e)苯乙烯基环丙烷基
‑
1,1
‑
二甲酸甲酯(ⅱa)(26.0mg,0.1mmol)和碘化镧(3.6mg,0.007mmol),然后向该反应瓶中加入色谱级乙酸乙酯(1.0ml),然后在油浴60℃下搅拌反应16h。反应结束后,将反应冷却至室温,然后用水(1.0ml)淬灭,再用二氯甲烷萃取(3x 4ml),将有机相用无水硫酸钠干燥,并用旋转蒸发仪将溶剂蒸干,得到的粗品再经柱色谱(洗脱剂为乙酸乙酯:二氯甲烷:石油醚=1:1:20,v/v/v)分离纯化得到白色固体3
‑
对甲苯磺酰亚胺基
‑
2'
‑
苯基
‑
5'
‑
(e)
‑
苯乙烯基
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
3',3'
‑
二甲酸甲酯(ⅲaa)58.5mg,收率92%。
[0044]
所得产物(ⅲaa)的表征数据如下:
[0045]
白色固体(58.5mg,92%);m.p.=178
‑
180℃;
[0046]1h nmr(400mhz,cdcl3)δ8.38(d,j=8.2hz,1h),7.99(d,j=8.2hz,2h),7.49(t,j=8.6hz,1h),7.38(d,j=8.0hz,2h),7.20
–
7.06(m,11h),6.91(t,j=7.7hz,1h),6.41(d,j=16.0hz,1h),5.90(dd,j=16.0,8.0hz,1h),4.79(s,1h),3.76(s,3h),3.51(t,j=13.8hz,1h),3.28
–
3.26(m,4h),2.54(dd,j=13.8,6.9hz,1h),2.48(s,3h);
[0047]
13
c nmr(100mhz,cdcl3)δ179.7,172.2,170.3,170.1,143.4,139.0,138.9,136.6,134.1,133.5,130.7,130.1,129.5,128.4,128.0,127.8,127.6,127.0,126.4,123.3,
122.2,118.5,112.2,101.7,63.3,60.4,53.2,52.7,52.3,39.0,21.6;
[0048]
hrms(esi):m/z calcd for c
37
h
34
no7s:636.2050[m h] ,found:636.2048。
[0049]
实施例2
[0050]3‑
对甲苯磺酰亚胺基
‑
2'
‑
苯基
‑
5'
‑
(2
‑
甲基丙烯
‑1‑
基)
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
3',3'
‑
二甲酸甲酯(ⅲab)的合成
[0051]
反应路线如下:
[0052][0053]
具体制备步骤如下:在手套箱中,氮气气氛下,向5ml圆底烧瓶中依次加入n
‑
((e)
‑2‑
((z)
‑
苯亚甲基)苯并呋喃
‑
3(2h)
‑
亚甲基)
‑4‑
甲基苯磺酰亚胺(ⅰa)(37.5mg,0.1mmol),(2
‑
甲基丙烯
‑1‑
基)环丙烷基
‑
1,1
‑
二甲酸甲酯(ⅱb)(21.2mg,0.1mmol)和碘化镧(3.6mg,0.007mmol),然后向该反应中加入色谱级乙酸乙酯(1.0ml),然后在油浴60℃下搅拌反应16h。反应结束后,将反应冷却至室温,然后用水(1.0ml)淬灭,再用二氯甲烷萃取(3x 4ml),将有机相用无水硫酸钠干燥,并用旋转蒸发仪将溶剂蒸干,得到的粗品再经柱色谱(洗脱剂为乙酸乙酯:二氯甲烷:石油醚=1:1:18,v/v/v)分离纯化得到白色固体3
‑
对甲苯磺酰亚胺基
‑
2'
‑
苯基
‑
5'
‑
(2
‑
甲基丙烯
‑1‑
基)
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
3',3'
‑
二甲酸甲酯(ⅲab)41.7mg,收率71%。
[0054]
所得产物(ⅲab)的表征数据如下:
[0055]
白色固体(41.7mg,71%yield);m.p.76
‑
78℃;
[0056]1h nmr(400mhz,cdcl3)δ8.37
–
8.34(m,1h),7.94(d,j=8.2hz,2h),7.54
–
7.48(m,1h),7.37(d,j=8.0hz,2h),7.13
–
7.11(m,2h),7.09(s,1h),7.07
–
7.04(m,3h),6.93(t,j=8.0hz,1h),4.96
–
4.89(m,1h),4.72(s,1h),3.73(s,3h),3.32
–
3.26(m,2h),3.24(s,3h),2.47(s,3h),2.39
–
2.37(m,1h),1.53(d,j=1.4hz,3h),1.47(d,j=1.3hz,3h);
[0057]
13
c nmr(100mhz,cdcl3)δ180.1,172.5,170.4,170.2,143.3,139.0,138.8,137.7,133.6,130.5,130.0,129.5,127.9,127.7,126.9,121.9,118.5,118.3,112.2,102.4,63.3,60.3,53.1,52.2,48.1,39.7,25.8,21.6,18.7;
[0058]
hrms(esi):m/z calcd for c
33
h
34
no7s:588.2050[m h] ,found:588.2047。
[0059]
实施例3
[0060]3‑
对甲苯磺酰亚胺基
‑
2'
‑
苯基
‑
5'
‑
(丁烯
‑2‑
基)
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
3',3'
‑
二甲酸乙酯(ⅲac)的合成
[0061]
反应路线如下:
[0062][0063]
具体制备步骤如下:在手套箱中,氮气气氛下,向5ml圆底烧瓶中依次加入n
‑
((e)
‑2‑
((z)
‑
苯亚甲基)苯并呋喃
‑
3(2h)
‑
亚甲基)
‑4‑
甲基苯磺酰亚胺(ⅰa)(37.5mg,0.1mmol),(丁烯
‑2‑
基)环丙烷基
‑
1,1
‑
二甲酸乙酯(ⅱc)(24.0mg,0.1mmol)和碘化镧(3.6mg,0.007mmol),然后向该反应中加入色谱级乙酸乙酯(1.0ml),然后在油浴60℃下搅拌反应16h。反应结束后,将反应冷却至室温,然后用水(1.0ml)淬灭,再用二氯甲烷萃取(3x 4ml),将有机相用无水硫酸钠干燥,并用旋转蒸发仪将溶剂蒸干,得到的的粗产品再经柱色谱(洗脱剂为乙酸乙酯:石油醚=1:10,v/v)分离纯化得到白色固体3
‑
对甲苯磺酰亚胺基
‑
2'
‑
苯基
‑
5'
‑
(丁烯
‑2‑
基)
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
3',3'
‑
二甲酸乙酯(ⅲac)46.2mg,收率75%。
[0064]
所得产物(ⅲac)的表征数据如下:
[0065]
白色固体(46.2mg,75%);m.p.=126
‑
128℃;
[0066]1h nmr(400mhz,cdcl3)δ8.38(d,j=8.1hz,1h),7.99(d,j=8.1hz,2h),7.50
–
7.44(m,1h),7.38(d,j=8.0hz,2h),7.18
–
7.11(m,2h),7.09
–
7.02(m,3h),7.00(d,j=8.4hz,1h),6.93(t,j=7.8hz,1h),5.04(s,1h),4.81(s,2h),4.30
–
4.22(m,1h),4.20
–
4.10(m,1h),3.92
–
3.84(m,1h),3.60(t,j=14.0hz,1h),3.54
–
3.41(m,1h),3.06(dd,j=14.3,6.3hz,1h),2.47(s,3h),2.41(dd,j=13.7,6.3hz,1h),1.85(q,j=7.4hz,2h),1.20(t,j=7.1hz,3h),0.82(d,j=7.3hz,3h),0.70(d,j=7.1hz,3h);
[0067]
13
c nmr(100mhz,cdcl3)δ180.2,171.8,170.3,169.8,144.7,143.3,139.0,138.8,133.7,130.5,130.3,129.5,127.8,127.6,126.9,121.9,118.5,112.9,112.3,100.7,62.6,62.0,61.4,60.6,53.8,37.9,28.8,21.6,13.9,13.2,12.4;
[0068]
hrms(esi):m/z calcd for c
35
h
38
no7s:616.2363[m c2h5] ,found:616.2363。
[0069]
实施例4
[0070]3‑
对甲苯磺酰亚胺基
‑
2'
‑
(4
‑
甲氧基)
‑
苯基
‑
5'
‑
乙烯基
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
3',2
”‑
茚二酮(ⅲbd)的合成
[0071]
反应路线如下:
[0072][0073]
式中,pmp为对甲氧基苯基。
[0074]
具体制备步骤如下:在手套箱中,氮气气氛下,向5ml圆底烧瓶中依次加入n
‑
((e)
‑2‑
((z)
‑
对甲氧基苯亚甲基)苯并呋喃
‑
3(2h)
‑
亚甲基)
‑4‑
甲基苯磺酰亚胺(ⅰb)(37.5mg,
0.1mmol),乙烯基环丙烷基
‑
1,1
‑
茚二酮(ⅱd)(19.8mg,0.1mmol)和碘化锂(6.7mg,0.05mmol),然后向该反应中加入色谱级乙酸乙酯(1.0ml),然后在油浴60℃下搅拌反应16h。反应结束后,将反应冷却至室温,然后用水(1.0ml)淬灭,再用二氯甲烷萃取(3x 4ml),将有机相用无水硫酸钠干燥,并用旋转蒸发仪将溶剂蒸干,得到的的粗产品再经柱色谱(洗脱剂为乙酸乙酯:二氯甲烷:石油醚=2:5:20,v/v/v)分离纯化得到白色固体3
‑
对甲苯磺酰亚胺基
‑
2'
‑
(4
‑
甲氧基)
‑
苯基
‑
5'
‑
乙烯基
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
3',2
”‑
茚二酮(ⅲbd)26.6mg,收率48%。
[0075]
所得产物(ⅲbd)的表征数据如下:
[0076]
白色泡沫状固体(26.6mg,48%);
[0077]1h nmr(400mhz,cdcl3)δ8.46(d,j=8.1hz,1h),8.08(d,j=8.2hz,2h),7.90(d,j=7.0hz,1h),7.81(d,j=7.0hz,1h),7.76
–
7.68(m,2h),7.50(t,j=7.8hz,1h),7.44(d,j=8.1hz,2h),7.08
–
6.99(m,2h),6.98(d,j=8.6hz,2h),6.45(d,j=8.8hz,2h),6.03
–
5.76(m,1h),5.11
–
4.96(m,2h),4.26(s,1h),4.20
–
4.08(m,1h),3.55(s,3h),2.50(s,3h),2.27(d,j=11.5hz,1h),2.19(dd,j=12.8,7.2hz,1h);
[0078]
13
c nmr(100mhz,cdcl3)δ202.3,201.1,182.0,170.6,158.9,143.4,142.1,141.5,139.1,138.9,136.0,135.6,134.7,131.7,130.5,129.5,126.9,124.0,123.3,123.1,122.1,118.2,117.5,113.3,112.4,99.9,64.5,54.9,54.1,38.2,21.7;
[0079]
hrms(esi):m/z calcd for c
36
h
30
no6s:604.1584[m h]
,found:604.1582。
[0080]
实施例5
[0081]3‑
对甲苯磺酰亚胺基
‑
2'
‑
甲基
‑
5'苯基
‑
2'
‑
乙烯基
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
4',4'
‑
二甲酸乙酯(ⅲaf)的合成
[0082]
反应路线如下:
[0083][0084]
具体制备步骤如下:在手套箱中,氮气气氛下,向5ml圆底烧瓶中依次加入n
‑
((e)
‑2‑
((z)
‑
苯亚甲基)苯并呋喃
‑
3(2h)
‑
亚甲基)
‑4‑
甲基苯磺酰亚胺(ⅰa)(37.5mg,0.1mmol),2
‑
甲基
‑
2乙烯基环丙烷基
‑
1,1
‑
二甲酸乙酯(ⅱf)(26.2mg,0.1mmol)和碘化锂(4.0mg,0.03mmol),然后向该反应中加入色谱级乙酸乙酯(1.0ml),然后在油浴60℃下搅拌反应16h。反应结束后,将反应冷却至室温,然后用水(1.0ml)淬灭,再用二氯甲烷萃取(3x 4ml),将有机相用无水硫酸钠干燥,并用旋转蒸发仪将溶剂蒸干,得到的的粗产品再经柱色谱(洗脱剂为乙酸乙酯:二氯甲烷:石油醚=1:1:16,v/v/v)分离纯化得到白色固体3
‑
对甲苯磺酰亚胺基
‑
2'
‑
甲基
‑
5'苯基
‑
2'
‑
乙烯基
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
4',4'
‑
二甲酸乙酯(ⅲaf)56.0mg,收率93%。
[0085]
所得产物(ⅲaf)的表征数据如下:
[0086]
白色泡沫状固体(56.0mg,收率93%);
[0087]1h nmr(400mhz,cdcl3)δ8.32(d,j=8.0hz,1h),7.80(d,j=8.2hz,2h),7.50(t,j=7.3hz,1h),7.28(d,j=8.1hz,2h),7.12
–
7.05(m,3h),6.99
–
6.96(m,3h),6.92(t,j=8.0hz,1h),6.18(dd,j=17.4,10.9hz,1h),4.99
–
4.87(m,2h),4.67(s,1h),4.16
–
4.03(m,2h),3.87
–
3.77(m,1h),3.48
–
3.35(m,2h),2.47(d,j=14.1hz,1h),2.41(s,3h),1.13(t,j=7.1hz,3h),0.94(s,3h),0.62(t,j=7.1hz,3h);
[0088]
13
c nmr(100mhz,cdcl3)δ176.8,172.2,169.8,169.6,143.3,139.0,138.9,138.8,134.0,130.5,130.3,129.4,127.9,127.6,126.8,122.0,118.8,114.1,112.1,102.7,62.6,61.9,61.6,57.7,53.8,44.7,21.6,18.9,13.9,13.2;
[0089]
hrms(esi):m/z calcd for c
34
h
36
no7s:602.2207[m h]
,found:602.2203。
[0090]
对比例1
[0091]3‑
对甲苯磺酰亚胺基
‑
2'
‑
苯基
‑
5'
‑
(2
‑
甲基丙烯
‑1‑
基)
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
3',3'
‑
二甲酸甲酯(ⅲab)的合成如实施例2所述,所不同的是:不加入催化剂,没有得到目标产物,原料几乎没有反应。
[0092]
对比例2
[0093]3‑
对甲苯磺酰亚胺基
‑
2'
‑
苯基
‑
5'
‑
(e)
‑
苯乙烯基
‑
3h
‑
螺[苯并呋喃
‑
2,1'
‑
环戊烷]基
‑
3',3'
‑
二甲酸甲酯(ⅲaa)的合成如实施例所述,所不同的是:反应温度为室温,所得产物收率为4.7%。
[0094]
本对比例中反应温度为室温,所得目标产物收率较低。
[0095]
以上仅是本发明的部分实施例而已,并非对本发明做任何形式上的限制,凡是依据发明的技术实质对上述实施例作的任何简单的修改,等同变化与修饰,均属于本发明技术方案范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。